不同酶处理对生物解离乳状液结构及稳定性的影响
2018-09-17齐宝坤谢凤英钟明明
李 杨 李 红 齐宝坤 谢凤英 钟明明 檀 政
(东北农业大学食品学院, 哈尔滨 150030)
0 引言
传统的大豆油脂制取方式主要有机械压榨法和溶剂浸出法。随着研究的深入,人们发现可以利用生物酶法(水酶法)提取大豆油脂,该方法是一种绿色环保的新技术。在提取过程中,含有酶制剂的水充当油料种子的提取媒介,在最适的酶解条件下反应,酶解后离心分离得到游离油、乳状液、水解液和残渣4部分[1]。由于处理时间过长,且乳状液难以分离,造成油脂无法被充分破除释放,因而限制了油脂提取率[2]。因此,如何采用高效、绿色、安全的破乳方法将乳状液中的油脂完全释放是提高生物解离油脂提取率的重要环节。
乳状液中加入酶能够将油脂分离出来,主要是由于加入到乳液的酶能水解界面蛋白和表面磷脂,从而减小其分子大小,减小油滴界面的刚性,进而释放被蛋白包裹的油脂。这些酶解反应可使油滴聚集变大,使得游离油更易释放[3-5]。JUNG等[6]研究报道,乳状液中加入LysoMax(磷脂酶A2)后破乳效果不好,这可能是由于形成了具有更强乳化性的溶血磷脂。磷脂酶D是大豆的一种内生磷脂酶,能将磷脂酰胆碱及磷脂酰乙醇胺两种大豆磷脂转变成磷脂酸。YAO等[7]研究表明,水酶法提取大豆油脂过程中,磷脂酰乙醇胺和磷脂酰胆碱向磷脂酸的转变与油脂游离油得率的降低有一定的相关性。
因此,本文采用2种蛋白酶和3种磷脂酶对乳状液进行处理,通过对粒径分布、Zate电位、显微镜观察、红外光谱以及荧光光谱的测定,探究不同生物酶处理大豆乳状液的蛋白质结构与稳定性之间的关系,明确乳状液体系中关键组分和空间构象以及组分相互作用对乳化相形成及稳定的影响机制。
1 材料与设备
1.1 材料与试剂
大豆,东北农业大学大豆研究所;碱性蛋白酶Protex 6L,杰能科生物工程有限公司;十二烷基硫酸钠(SDS)、β-巯基乙醇、磷酸氢二钠、磷酸二氢钠盐酸、考马斯亮蓝(G-250)、牛血清蛋白(BSA)、氢氧化钠、丙酮、无水乙醇等化学试剂均为分析纯级。
1.2 仪器与设备
FW100型高速万能粉碎机,绍兴科宏仪器有限公司;挤压膨化机,东北农业大学自制;BX53型正置生物显微镜,OLYMPUS公司;LGJ-1型冷冻干燥机,上海医用离心机厂;GL-21M型高速冷冻离心机,上海市离心机械研究所; TNZ1-5700型傅里叶红外光谱仪,英国Thermo Fisher公司;F2000型荧光光谱仪,日本Hitachi公司;Mastersizer 2000型粒度仪,英国马尔文仪器有限公司。
2 试验方法
2.1 生物解离大豆乳状液的制备
参照齐宝坤[8]的方法,并作适当的修改,大豆粉碎后挤压膨化预处理,得到膨化大豆粉。膨化大豆粉与水混合(液料比6 mL/g),加入碱性蛋白酶Protex 6L,搅拌均匀后在60℃水浴锅中保持恒温加热,用2 mol/L NaOH调节pH值至9.0,酶解3 h后,取出于100℃沸水中灭酶10 min,在4 500 r/min转速下离心20 min,离心后吸取游离油,将其余液体部分倒入分液漏斗,静置24 h分层,将乳状液分离。
2.2 乳状液的生物酶处理
分别采用蛋白酶和磷脂酶处理乳状液[9-10],每种酶在其最适的处理条件,酶解反应时间均为1 h,酶的添加量为2%(酶与乳状液质量百分比)。碱性蛋白酶Protex 6L和碱性蛋白酶Acalase 2.4L在pH值为8、温度为40℃条件下进行酶解;溶血磷脂酶在pH值为4.5、温度为40℃进行酶解;磷脂酶A2和磷脂酶D在pH值为8、温度为40℃条件下进行酶解,酶解后在3 585g下离心10 min,来分离乳状液和水相。
2.3 乳状液分层系数和游离油得率的测定
分层系数(Creaming index)的测定:取8 mL处理后的乳状液于离心管中,在4℃贮藏24 h,乳状液发生分层现象,上相浑浊,下相澄清。分层系数计算公式为
式中Hs——下相的高度
Hr——乳状液总高度
游离油得率计算公式为
式中m——破乳后总游离油质量
M——乳状液中含油质量
2.4 乳状液粒径及Zate电位的测定
采用激光光散射粒度分析仪测定乳状液体积加权平均值(D4,3),将小部分乳状液分散在50 mL蒸馏水中,保持粒径数值在测定范围内,大豆油滴的折射指数为1.47,分散剂的折射指数为1.333。测定在室温(20℃)下进行,吸光度为0.001。
采用Zetasizer Nanozs 90型电位仪测定乳状液溶液的Zate电位。0.05 mol/L pH值7.0的磷酸盐缓冲溶液将大豆乳状液样品稀释至质量分数为0.2%的溶液后进行测定,上样体积1 mL,测定温度25℃。每个样品重复测量3次。
2.5 乳状液正置显微镜分析
利用配有DP27型显微数码相机的BX53型正置生物显微镜对乳状液的显微结构进行观察。用吸管取2~3滴乳状液于干净干燥的载玻片上,轻轻地加上盖玻片后,置于显微镜明场放大100倍进行观察。
2.6 乳状液中蛋白质的提取
乳状液水相中蛋白质提取方法采用丙酮沉淀法[11],将20 g/mL冰丙酮在-18℃下反应2 h,离心分离(12 000g,15 min,4℃),除去上清液,将沉淀继续用冰丙酮洗涤4~5次,直到溶剂由黄色变成无色,沉淀中溶剂挥发后冻干得到蛋白,放入4℃冰箱备用。
2.7 表面疏水性的测定
依据LAEMMLI[12]的方法,即ANS(8-苯氨基-1-萘磺酸铵盐)荧光探针法。称取0.025 g不同处理方式下的蛋白样品溶于50 mL磷酸盐缓冲液中,配成pH值为7、0.01 mol/L的溶液,将溶液在室温下搅拌混合1 h,然后在10 000g下离心处理30 min,取上清液采用Lowry法测定其蛋白质浓度,并用上述磷酸盐缓冲溶液依次稀释后,使其浓度在0.005~0.5 mol/mL之间,取不同浓度样品的溶液4 mL,分别加入40 μL浓度为8 mmol/L的ANS溶液,经振荡混合后静置5 min,再测定样品的荧光强度。试验中激发波长为370 nm,发射波长为490 nm,夹缝宽为5 nm。
2.8 红外光谱的测定
将样品粉碎后过100目的筛子,粉末在40℃干燥箱中干燥12 h,然后在红外灯下进行研磨处理。称取约2 mg的待测样品,加入200 mg溴化钾,在玛瑙研钵中研磨15 min,随后进行压片处理,压片机在140 N压力下约保持1 min,然后将制得的均匀透明薄片放入红外光谱仪中进行测定。测定的条件为:扫描范围400~4 000 cm-1,分辨率为4 cm-1,扫描信号累加64次。每种处理的图谱扫描重复3次。
谱图的分析处理采用Peakfit软件,在酰胺Ⅰ带1 600~1 700 cm-1进行两点基线校正,然后再采用Savitsk-Golay函数进行平滑处理,求二阶导数谱,并采用Gauss峰形进行拟合,然后估算出子峰的个数与位置,经过手动调整各子峰的峰高和半峰宽,进行多次拟合使得残差最小,确定各子峰与各二级结构的对应关系后,根据其积分面积就可以计算出4种二级结构的相对含量。
2.9 荧光光谱的测定
依据尹寿伟[13]的方法,采用F-4500型荧光分光光度计测定乳状液分离出的大豆蛋白色氨酸荧光光谱。将大豆蛋白样品分别分散于0.01 mol/L、pH值为7.0的磷酸缓冲液中,配制蛋白质量浓度为0.15 mg/mL的溶液。荧光发散光谱分析以蛋白质分子内部的色氨酸荧光基团为探针,为了降低酪氨酸的贡献,荧光光谱的激发波长为290 nm,光谱的扫描范围为300~400 nm,激发与发射狭缝的宽均为5 nm。
2.10 乳状液中磷脂的提取与测定
乳状液中的磷脂提取方法如下[14]:
(1)粗油提取:向乳状液中加入200 mL甲醇,混合摇匀后加入400 mL氯仿继续混匀后,在室温下磁力搅拌4 h,然后采用布式漏斗回收固体和萃取液,将固体反复萃取,将得到的萃取液合并,萃取液经过旋转蒸发除去溶剂,继续用氯仿、甲醇、0.74%水溶性KCl按照体积比8∶4∶3进行洗涤,最终的粗油储存在-26℃下备用。
(2)磷脂浓缩:将100 mL正己烷与100 mL体积分数为87%的乙醇混合,混合后放入分液漏斗中,使之静置平衡,静置后分离得到上层正己烷相(溶剂A)和较低的乙醇相(溶剂B);约10 g粗油在200 mL分液漏斗中与45 mL溶剂A和15 mL溶剂B混合,5 min后相达到平衡,收集较低的乙醇相,15 mL溶剂B加入分液漏斗上层相中,相平衡后将较低的乙醇相收集与第1次得到的乙醇相合并,此过程重复10次,乙醇提取物与130 mL氯仿和0.1 mol/L K-EDTA(111 mL,pH值7)混合,然后收集较低的氯仿相,采用硫酸钠干燥,除去溶剂的浓缩油在-26℃储存待用。
乳状液磷脂测定采用31P-NMR(磷-31核磁共振)分析,浓缩的粗油(80~90 mg)加入TPP(磷酸三苯酯,10 mg,固体)溶解在氯仿、甲醇Cs-EDTA组成混合液中,剧烈震荡摇匀,离心后将较低的部分转移到5 mm NMR瓶中进行测定。31P-NMR测定条件如下:探头温度29℃,脉冲宽度22 μs,扫描宽度9 718 Hz,采集时间1、2 s,间隔10 s,扫描次数256次。
3 结果与讨论
3.1 分层系数和游离油得率
分别采用碱性蛋白酶Protex 6L、碱性蛋白酶Acalase 2.4L、溶血磷脂酶、磷脂酶A2和磷脂酶D处理的乳状液,乳状液的分层系数和游离油得率如图1所示,碱性蛋白酶Protex 6L、碱性蛋白酶Acalase 2.4L分别在pH值为8的条件下进行水解,与原始乳液相比,其分层系数和游离油得率显著增加,游离油得率分别达到90.05%和94.24%,油脂得率的增加可能是由于采用蛋白酶处理乳状液时,蛋白酶能水解蛋白生成短肽,进而破坏了乳状液界面的完整性,使得其不再具有足够力量阻止油脂的聚集,油脂得以释放[15-16]。采用磷脂酶处理后游离油得率均增加,有研究表明磷脂酶处理水解包裹在油脂表面的磷脂,降低乳状液的稳定性,进而释放游离油[17]。溶血磷脂酶处理效果较好,游离油得率都可达到95.12%左右,可能由于溶血磷脂酶处理的pH值为4.5,采用磷脂酶D和磷脂酶A2处理的pH值为8,磷脂酶A2处理后游离油得率较前两种酶有所降低,为86.67%左右。本研究说明大豆蛋白和磷脂在液滴的界面上对乳状液的稳定性起着很重要作用,这与JUNG等[18]研究结果相一致。
3.2 乳状液稳定性
图2和图3所示为不同种类的生物酶对形成乳状液处理后的Zeta电位和粒径分布的影响。原始乳状液的Zeta电位值为(-21.57±1.88) mV,经两种碱性蛋白酶Protex 6L和Acalase 2.4L处理后乳状液的负电位绝对值降低,乳状液粒径分布主要集中在50~100 μm,可能的原因是经由蛋白酶水解后使水解物分子量变小并水解成小分子的多肽[19],因而会导致在油滴表面的吸附量降低,再重新与磷脂复合吸附在油-水界面,所形成的膜强度就会下降;另外一方面,蛋白的过度水解使表面电荷减少、空间位阻的作用下降,也会造成了大量油滴的聚集,使乳液的粒径增大[20]。磷脂酶A2酶解的乳状液负电位绝对值较大,粒径较小,分布范围较窄,大粒径液滴数量减少,乳化体系相对较为稳定,说明此时表面所带的负电荷较多,乳状液中磷脂非极性基团数量减少,亲水性基团增强,使表面的蛋白与酶解后的磷脂相互作用增强,较好地吸附在油水界面,进而使乳液的粒径变小[21],这与CHEN等[22]的研究结果相类似。经溶血磷脂酶酶解后其电位值为(-4.8±0.6) mV,出现较大部分粒径的分布,而经磷脂酶D酶解的乳状液电位变化到(-11.8±0.8) mV,这可能是由于磷脂酶D作用后使磷脂的结构发生改变甚至被破坏,使乳状液表面所带电荷发生变化,但此时表面电荷密度不大,液滴之间的静电斥力也相对较小[23],溶血磷脂酶和磷脂酶D主要作用磷脂的疏水尾部[24],由于被蛋白包裹乳液界面的磷脂被水解的程度相对较弱,但可能会造成磷脂结构略微不稳定,形成了较大的乳滴。
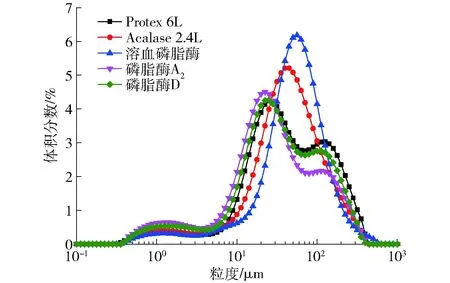
图3 不同酶处理对乳状液粒径分布的影响Fig.3 Effect of different enzymes treatments on particle size distribution of oil emulsion
3.3 乳状液微观结构
不同酶处理后乳液的微观结构采用光学显微镜进行观察,如图4所示。不同酶进行酶解处理乳状液呈现不同的形态。从图4可看出,经Protex 6L和Acalase 2.4L两种碱性蛋白酶处理后的乳状液微观结构相似,出现大尺度油滴的聚集;溶血磷脂酶酶解后的体系乳状液液滴中出现了油滴的成片聚集,乳状液界面出现了破裂,且液滴形状不规则、大小不均一,溶血磷脂酶水解后乳状液界面上磷脂被水解,界面强度下降,乳液极不稳定,随着油滴的聚集,碰撞使油滴之间出现聚结,从而使液滴粒径进一步增大,导致出现有较多的大油滴,此外,在pH值为4.5的条件下,乳状液中油滴也会相互聚集。磷脂酶D与碱性蛋白酶处理后的乳液分布状态相似,油滴的聚集和粒径的分布介于中间,磷脂酶A2酶解后的体系乳状液系中乳滴较小,可能是由于乳液界面磷脂部分水解形成的水解产物与蛋白重新乳化后表面带有更多电荷,具有抵抗乳液液滴聚集的能力[25],说明磷脂酶A2对乳状液破除效果不好。由光学显微镜观察来看,酶解后乳状液粒径、油滴的分布大小差别较大,表明油滴之间发生聚集现象,其乳状液形态与粒径大小测定结果一致。

图4 不同酶酶解对乳状液微观结构的影响Fig.4 Effects of different enzymes treatment on microscopic structure of emulsion
3.4 乳状液中蛋白质分析
3.4.1乳状液表面疏水性
经不同酶酶解后乳状液界面蛋白的表面疏水性指数是用来表征酶解后蛋白表面疏水基团数量的一个重要指数,也与蛋白的乳化特性有关,蛋白质的表面疏水性指数越高,其乳化能力越强[26]。图5为不同酶酶解后乳状液界面蛋白的表面疏水性变化,乳状液界面的磷脂和蛋白在酶作用下,会影响界面蛋白的表面疏水性,从而影响相互作用及界面强度。经Protex 6L和Acalase 2.4L两种碱性蛋白酶处理后的乳状液界面蛋白表面疏水性与原始乳液相比都呈现下降,主要是由于蛋白酶会影响界面吸附蛋白的分子结构,导致游离的疏水基团裂解或者暴露出的疏水基团发生聚集,引起水解后蛋白表面疏水性的降低。其中Acalase 2.4L处理的表面疏水性最小,说明此时蛋白水解物中有更多的亲水基团暴露,另外蛋白疏水性的变化会对蛋白的乳化性有重要影响,蛋白酶的过度酶解使得界面维持蛋白的内部结构力(包括氢键、范德华力、离子键等)逐渐被破坏,使油滴表面的保护层越来越薄,导致了乳化性的降低[27]。而用磷脂酶A2和磷脂酶D处理后的蛋白表面疏水性降低不明显,可能是由于磷脂经酶解后头部结构被破坏[28],但疏水的尾部并没有暴露,与蛋白相互作用时,被紧密包埋在区域内,导致疏水性没有明显增加。而采用溶血磷脂酶处理的乳状液,由于pH值的作用,其表面疏水性明显降低。
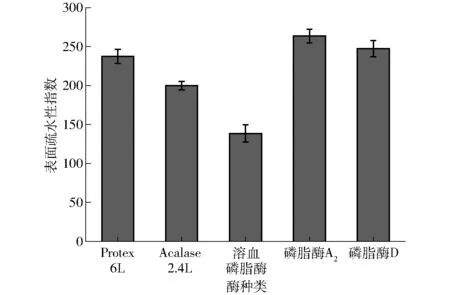
图5 不同酶酶解后乳状液蛋白的表面疏水性Fig.5 Surface hydrophobicity of oil emulsion with different enzyme treatments
3.4.2乳状液中蛋白质红外光谱分析

图6 不同酶处理条件下乳状液中蛋白质的红外光谱图Fig.6 FTIR of protein in oil emulsion with different enzymes treatment
乳状液中的蛋白二级结构组成与蛋白分子的空间构象密切相关,因而会影响蛋白的功能特性。红外光谱可显示蛋白质酰胺Ⅰ带、酰胺Ⅱ带、酰胺Ⅲ带等蛋白质结构中的相关信息[29]。傅里叶变换红外光谱的研究可以定量给出每组水解物中蛋白质的二级结构含量。图6为不同酶处理条件下乳状液中蛋白质的红外光谱图。研究表明,蛋白质α-螺旋二级结构对应波数为1 646~1 664 cm-1;β-折叠结构对应波数为1 615~1 637 cm-1和1 682~1 700 cm-1;β-转角结构对应波数为1 664~1 681 cm-1;无规卷曲结构对应波数为1 637~1 645 cm-1[30]。采用Gauss面积法拟合,通过峰位归属确定不同酶解物的二级结构种类和含量,计算结果见表1。原始乳状液的二级结构组成为:α-螺旋相对含量(17.30±0.10)%,β-折叠相对含量(40.90±0.18)%,β-转角相对含量(18.40±0.07)%,无规卷曲相对含量(23.40±0.12)%。由表1可知,其中酶解后的乳状液中蛋白的二级结构主要以β-折叠和无规卷曲为主,Protex 6L和Acalase 2.4L碱性蛋白酶酶解后,蛋白质中α-螺旋相对含量降低,分别降到(14.58±1.18)%和(10.42±1.28)%,无规卷曲结构相对含量升高。这可能是由于α-螺旋和β-折叠属于相对规则的构象,而β-转角、无规卷曲的蛋白质分子结构比较疏松,经酶解后蛋白质的肽链变得伸展,柔韧性增加,疏水性基团暴露,致使在乳状液中蛋白质二级结构相对含量不同,有利于吸附在油滴表面[31]。采用溶血磷脂酶、磷脂酶A2和磷脂酶D处理后的蛋白质α-螺旋相对含量均降低,无规卷曲相对含量升高,所发生的变化可能是由于酶解的磷脂水解物与蛋白分子发生交互作用时,部分影响了二级结构的转移。其中溶血磷脂酶的无规卷曲相对含量达到(32.40±0.15)%,反应pH值4.5使得蛋白发生变性,蛋白结构部分或者全部打开,导致有序结构含量降低,无序结构含量增加。

表1 不同酶处理条件下乳状液中蛋白质的二级结构相对含量Tab.1 Secondary structure content of protein in oil emulsion with different enzymes treatment %
注:表中同一列不同字母表示数值存在显著差异(P<0.05)。
3.4.3乳状液的内源荧光光谱分析
由图7可知,与原始乳状液荧光强度(377.82±6.86)相比,所有样品经碱性蛋白酶和磷脂酶酶解处理后,荧光强度都降低。在溶血磷脂酶酶解后乳状液中蛋白质的荧光强度最低,这可能与溶血磷脂酶的酶解pH值为4.5有关。经Protex 6L和Acalase 2.4L两种碱性蛋白酶处理后的乳状液中蛋白质最大吸收波长和荧光强度降低,并且乳状液荧光峰发生红移,可能是经蛋白酶酶解后蛋白结构不稳定,易发生基团移位[32],说明经蛋白酶酶解后界面蛋白的高级结构被破坏,肽键断裂,水解产物的亲水性显著增加,原来处于蛋白质分子内部的色氨酸逐渐暴露于水环境中,其氨基酸残基所处微环境的极性也逐渐增强,使其疏水性基团的数目减少,不能吸附在油滴表面,进而不能形成界面蛋白膜,而且红移的程度反映蛋白质构象变化的程度[33],这一结论与表面疏水性数据结果的分析一致。经溶血磷脂酶和磷脂酶D两种磷脂酶处理导致了光谱的改变,关于不同磷脂酶酶解的产物结构及其不稳定机理有待进一步研究。
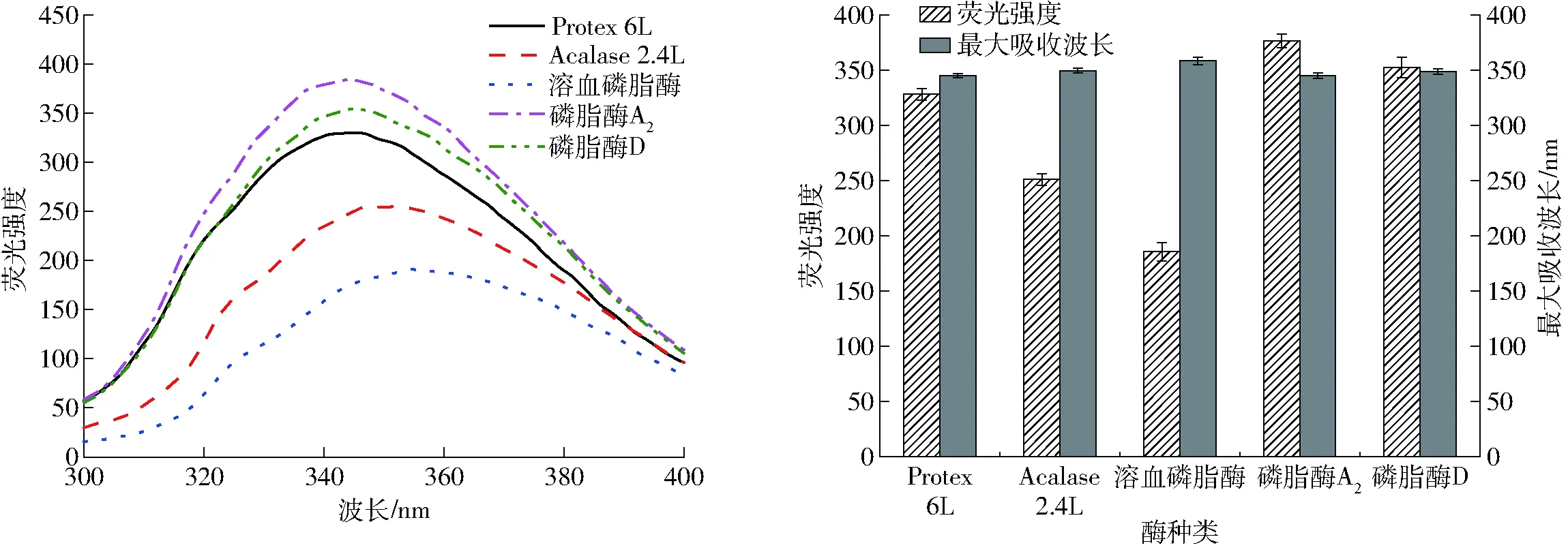
图7 不同酶处理条件下乳状液中蛋白质的内源荧光光谱分析Fig.7 Intrinsic fluorescence spectra analysis of protein in oil emulsion with different enzymes treatment
3.5 磷脂酶对乳状液磷脂的影响
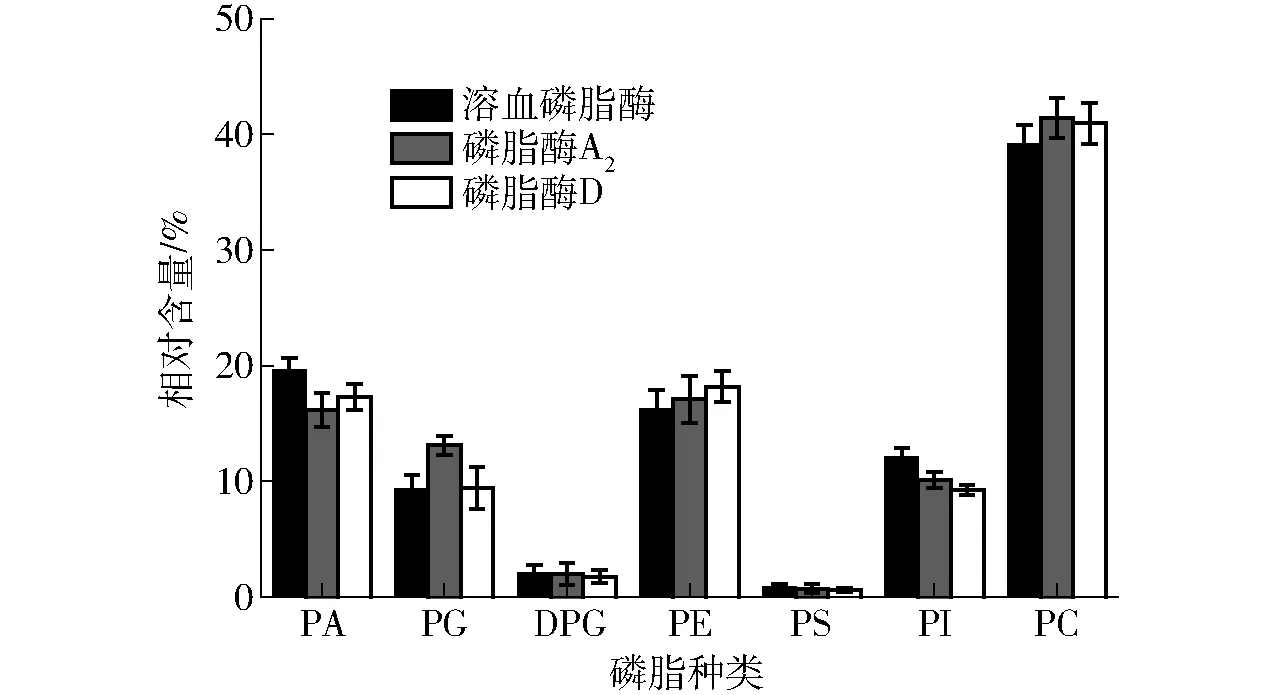
图8 不同酶酶解乳状液后磷脂含量31P-NMR图谱Fig.8 31P-NMR spectrum of phospholipids from oil emulsion with different enzymes treatment
如图8所示,本研究表明溶血磷脂酶可以切断酰基磷酸甘油酯的Sn-1键产生游离脂肪酸和甘油磷脂酰胺。磷脂酶A2水解磷脂分子中的Sn-2位置得到溶血磷脂和脂肪酸。而磷脂酶D则切断Sn-4位置的键。MENESES等[34]研究发现,磷脂酰胆碱(PC)为大豆粗油中含量最多,且化学位移最不稳定的。原始乳状液中磷脂主要有磷脂酰胆碱(PC)、磷脂酰乙醇胺(PE)、磷脂酰肌醇(PI)、磷脂酸(PA)、磷脂酰甘油 (PG)、磷脂酰丝氨酸(PS)和双磷脂酰甘油(DPG),相对含量分别为44.12%、16.4%、11.24%、15.6%、16.4%、0.23%、0.88%。由图8可知,采用溶血磷脂酶、磷脂酶A2和磷脂酶D酶解乳状液后,PA含量均升高,PC和PE降低,采用溶血磷脂酶酶解后PA相对含量达到(19.6±1.21)%,比原始乳状液增长了接近4个百分点,而采用磷脂酶A2和磷脂酶D处理乳状液后,PA相对含量也分别达到(16.2±1.45)%和(17.3±1.08)%。磷脂酶水解酰基磷酸甘油酯生成磷脂酸和溶血磷脂,使得PA含量升高。采用溶血磷脂酶、磷脂酶A2和磷脂酶D处理乳状液后,PC相对含量分别降低到(39.12±1.66)%、(41.38±1.73)%和(40.96±1.79)%。PS和DPG含量均升高,但3种酶处理变化不显著。采用磷脂酶A2处理后的PG含量最高,而溶血磷脂酶处理后的PI含量最高。此外,研究了磷脂酶D,因为它在处理过程中涉及形成不可水解的磷脂,即磷脂酸和溶血磷脂酸,其可影响大豆油质量[35]。
4 结论
(1)采用蛋白酶处理乳状液时,蛋白酶能水解蛋白生成短肽,分子量变小,表面疏水性降低,蛋白二级结构发生改变,荧光强度降低,进而破坏了乳状液界面蛋白的完整性,导致在油滴表面的吸附量降低,失去部分电荷,使其不再具有足够力量阻止油脂的聚集,油滴粒径增大,解决了生物酶法提取大豆油脂处理时间过长、无法充分破除释放油脂的问题,油脂提取率得以增加。可以利用这种高效的破乳技术,获得高提取率的大豆油脂,为生物解离提取大豆油脂产业化提供理论依据。
(2)磷脂酶处理水解了包裹在油脂表面的磷脂,PC和PE水解,PA含量均升高,溶血磷脂、磷脂酶A2酶解Sn-1和Sn-2位后,使磷脂头部负电性增强,表面所带的负电荷较多,磷脂酶D的酶解位点为Sn-4位,由于Sn-4位距离磷脂头部较近,将磷脂头部呈负电性的磷酸基团酶解成游离的脂肪酸,降低乳状液的稳定性,油滴的粒径增大,进而增加了游离油得率。
