整合GWAS和WGCNA分析挖掘甘蓝型油菜黄籽微效作用位点
2018-08-10鲜小华徐新福曲存民李加纳刘列钊
鲜小华 王 嘉 徐新福 曲存民 卢 坤 李加纳 刘列钊,*
整合GWAS和WGCNA分析挖掘甘蓝型油菜黄籽微效作用位点
鲜小华1,**王 嘉1,2,**徐新福1曲存民1卢 坤1李加纳1刘列钊1,*
1西南大学农学与生物科技学院 / 西南大学农业科学研究院, 重庆 400715;2南充市农业科学院, 四川南充 637000
甘蓝型油菜是世界上最重要的油料作物之一, 黄籽是提高品质的重要育种目标。本研究以520份具有代表性的甘蓝型油菜品种(系)为材料, 结合种子发育过程中8个时期的转录组数据, 采取整合全基因组关联分析(GWAS)和权重基因共表达网络分析(WGCNA)的策略, 挖掘油菜黄籽性状微效作用位点, 2年共检测到199个SNP位点, 在SNP位点附近共挖掘出1826个名义候选基因。利用R语言中的WGCNA软件包构建了8个共表达模块, 基因功能富集分析显示, turquoise模块和blue模块与黄籽表型相关。苯丙烷代谢途径、类黄酮途径的关键基因、以及为turquoise模块的枢纽基因(hub gene)。通过已知的黄籽相关基因, 挖掘出了一部分黄籽微效作用基因, 这些基因多参与苯丙烷、类黄酮以及原花青素代谢途径。本研究挖掘的这些位点和候选基因可作为影响油菜黄籽形成的重要候选区域和基因, 有助于探究甘蓝型油菜黄籽基因资源信息、揭示油菜黄籽性状的遗传基础和分子机制、丰富分子育种理论以及提高油菜品质。
甘蓝型油菜; 黄籽; 全基因组关联分析; 权重基因共表达网络分析
甘蓝型油菜(L.)是全世界广泛种植的主要油料作物之一, 同时也是我国重要的油料、饲料和能源作物。黄籽是甘蓝型油菜的一个重要性状, 在相同的遗传背景下, 甘蓝型黄籽油菜比黑籽油菜具有更高的含油量和蛋白质含量[1]。同时, 黄籽油菜种皮薄, 籽粒纤维含量低, 对饲料品质更为有利; 其油质清澈, 原油中色素含量少, 便于脱色加工[2]。因而对黄籽油菜的遗传解析一直是油菜品质育种的重要工作之一。甘蓝型油菜种皮色泽不仅表现有纯黑色和纯黄色, 还表现出中间不同类型的颜色, 如红色、红褐色、黑褐色等, 表明种皮色泽不仅受主效位点控制, 同时也受微效多基因影响。
基于连锁不平衡的全基因组关联分析(genome- wide association study, GWAS), 又称作关联作图或连锁不平衡作图, 其原理是利用自然群体中存在的丰富变异, 使用特定的统计方法寻找出与目标性状变异显著相关的位点或基因组区段[3]。随着生物信息学的迅猛发展以及测序成本的下降, GWAS被广泛应用于解析动植物复杂数量性状的研究中。自Hasan等[4]首次将关联分析应用于甘蓝型油菜种子硫苷含量的研究中以来, GWAS分析越来越频繁地出现在甘蓝型油菜重要数量性状的遗传研究中。在前期研究中, 我们对520份材料进行GWAS分析, 2年共检测到22个SNP位点与黄籽表型显著关联, 挖掘出14个与黄籽形成相关的基因[5]。与传统的连锁作图相比较, GWAS具有耗时少、广度大、精确度高等优势[6], 但也存在一定的局限性, 其面临的最大的问题之一是其对微效多基因控制的数量性状检测能力不足, 难以准确挖掘出微效作用位点, 产生遗传性缺失现象(missing heritability)。
同样是基于基因与性状之间的关联分析技术, 权重基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)更加注重挖掘和呈现基因在不同样本中的表达形式, 基于表达模式类似的分子可能参与特定生物学功能的理论[7], 利用基因表达数据构建生物体内基因间的潜在作用网络体系, 以探索基因网络与性状之间的关联关系。自该技术问世以来, 因其强大的分析效能, 在生物医学研究中得到广泛应用[8], 随后迅速扩大到多个研究领域并被广泛运用。基于WGCNA能够对复杂的数据降维并简化为若干模块, Weiss等[9]提出基因模块相关性研究(gene module association study, GMAS)作为GWAS分析结果的补充。Farber[10]整合GWAS和WGCNA分析, 并成功发掘出在常规GWAS分析中没有被发现的与骨密度相关的基因, 同时验证了基因在骨代谢中的核心地位。该方法显著提高了对微效基因的挖掘效率, 为GWAS解决自身的局限性找到了捷径。本研究首次采用整合GWAS和WGCNA分析的策略, 对甘蓝型油菜黄籽表型进行遗传分析, 挖掘控制黄籽表型的相关作用位点, 旨在为甘蓝型黄籽油菜育种研究提供有价值的信息和依据。
1 材料与方法
1.1 材料与数据来源
用于GWAS分析的群体由不同育成年代、不同生态区域的国内外520份材料组成。在前期的研究中, 我们利用该群体已完成对黄籽R-value的GWAS分析, 本研究所使用的数据来源于前期发表的GWAS分析结果, 使用的甘蓝型油菜种子发育和成熟过程中的基因表达谱数据来自NCBI数据库中的GEO数据库, 其注册码为GSE43918[11]。该数据包含8个取样时间点(分别为开花后第10、第15、第20、第25、第30、第35、第40以及第45天), 每个时间点6个重复共计48个样本。
1.2 名义显著关联基因及表达谱数据的获得
采用相对宽松的5% FDR对GWAS分析结果进行多重校正, 获得2014年和2015年名义上与黄籽性状相关联的基因集。利用BLAST, 将表达谱数据中的探针序列与候选基因比对, 筛选无错配、无缺失的高质量比对结果, 获得该基因集的表达谱数据, 对于多个探针比对到同一个基因的情况, 取平均值作为该基因的表达数据。
1.3 加权基因共表达网络构建
利用R软件的WGCNA包完成共表达网络构建, 参考WGCNA官网(https://labs.genetics.ucla.edu/ horvath/CoexpressionNetwork/Rpackages/WGCNA/)上的tutorial完成[12]。首先, 对样品聚类分析, 去除离群样本; 为了尽量满足无尺度网络分布前提条件, 利用WGCNA提供的pickSoftThreshold函数计算软阈值, 选取拟合曲线第一次接近0.9时的阈值参数β; 根据β值, 计算所有基因间的相关性创建邻接矩阵, 将邻接矩阵转换为拓扑矩阵, 利用相异度进行拓扑重叠, 采取逐步法构建基因联系网络。利用动态剪枝法识别共表达模块, 设定每个模块最低基因数为30个。计算每个模块的特征向量基因ME (Module Eigengene)对模块聚类作降维处理, 选择相似性在75%以上模块进行融合(MEDissThres=0.25)。
1.4 黄籽相关模块的查找、分析及枢纽基因(hub gene)的筛选
利用OS-tools的GO分析软件分别对各模块内的基因进行富集分析, 选择甘蓝型油菜基因组作为背景, 以校正后FDR < 0.05作为显著性阈值。利用相关系数法计算每个模块特征值与性状的相关性, 基于KEGG生物学通路数据库(http://www.kegg.jp/), 对黄籽表型特异相关的模块内的基因进行通路富集分析。利用OS-tools网络图工具完成基因间的作用关系可视化展示。
2 结果与分析
2.1 低显著性关联基因的挖掘及表达谱数据的获得
如图1所示, 在前期的研究中, 我们采用基于FWER标准的Bonferroni校正法对GWAS分析结果进行多重校正, 2年共检测到22个显著关联位点[5]。为获得更多微效关联位点, 本研究采用相对宽松的FDR校正法进行多重校正, 经校正, 2年共获得199个显著位点。根据该关联群体的LD衰减距离, 在显著位点上下游各50 kb内的区域共挖掘出1826个候选基因。利用从NCBI数据库中获得的转录组数据探针序列, 与候选基因比对, 去除低质量的比对结果, 共得到1461个低显著性关联基因的表达谱数据用于WGCNA分析。
2.2 共表达网络构建及模块筛选
利用R中的WGCNA软件包对低显著性关联基因集进行加权基因共表达分析, 采取逐步法构建共表达网络, 利用动态剪枝法识别共表达模块, 共获得8个模块(表1和图2-A), grey模块代表未能分配到任何一个模块的基因, 各模块所包含基因数目差异较大, 最少的pink模块仅包含49个基因, 最多的turquoise模块则含有473个基因。对8个模块分别进行GO (gene ontology)功能富集分析, 其中有4个模块没有富集到任何GO术语(GO term), 剩余4个模块共富集到189个GO term, 最显著的term是turquoise模块中的苯丙烷生物合成过程(GO: 0009699, FDR=1.47E–04)。4个模块中, turquoise模块共富集到156个GO term, 其中多个GO term与原花青素、木质素以及类黄酮生物合成相关; blue模块富集到6个GO term, 其中4个与木质素的生物合成相关(GO:0045551、GO:0052747、GO: 0009698和GO:0009699)。同时, 样本标量矩阵与模块特征向量相关性分析结果显示, turquoise模块有最高的相关性(= 0.63,= 2E–05), 其次为blue模块(图2-B)。
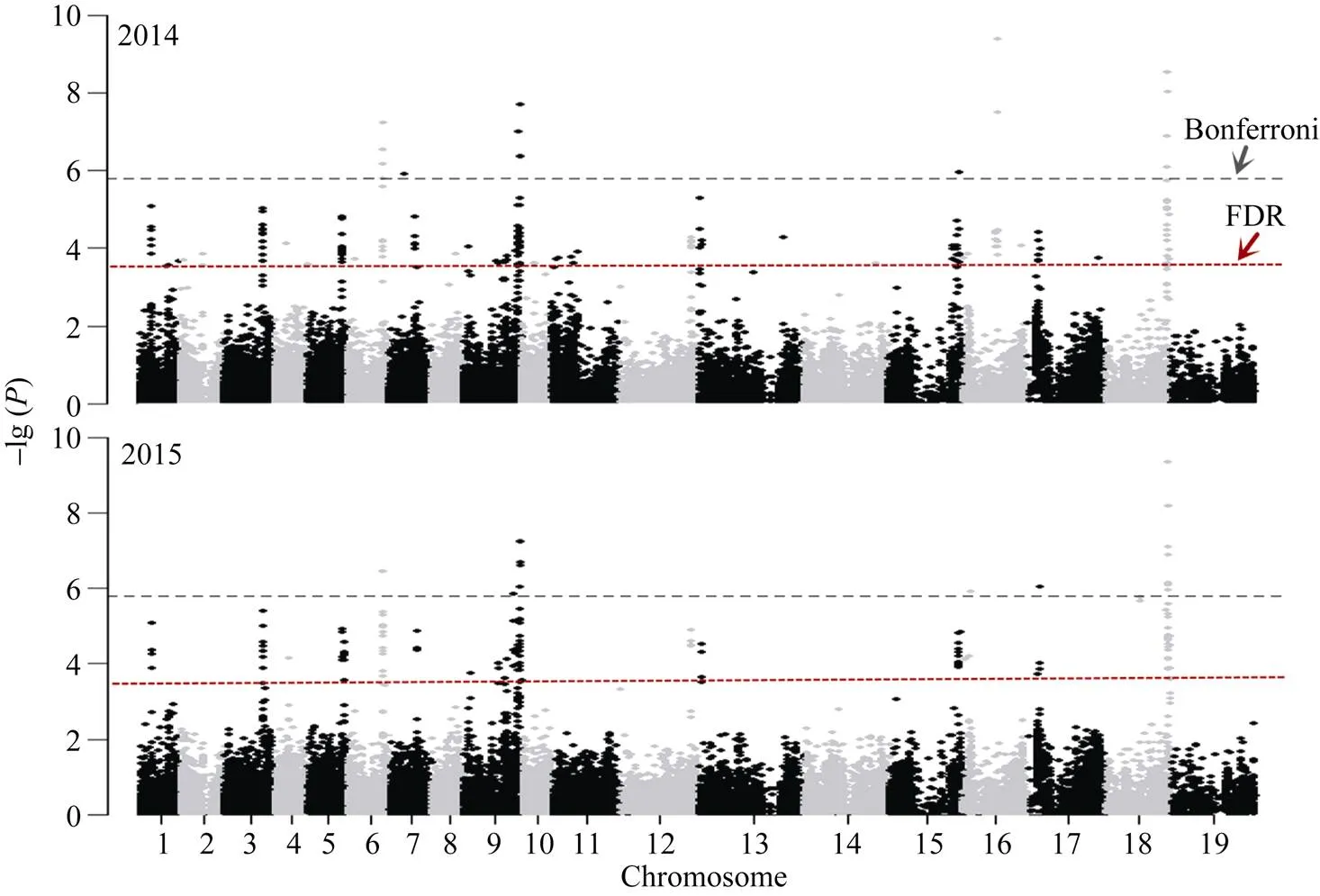
图1 甘蓝型油菜黄籽表型全基因组关联分析结果的曼哈顿图
灰色的虚线为Bonferroni校正阈值线, 红色为FDR校正阈值线。
The gray horizontal dashed line indicates the Bonferroni threshold; the red horizontal dashed line indicates the FDR threshold.
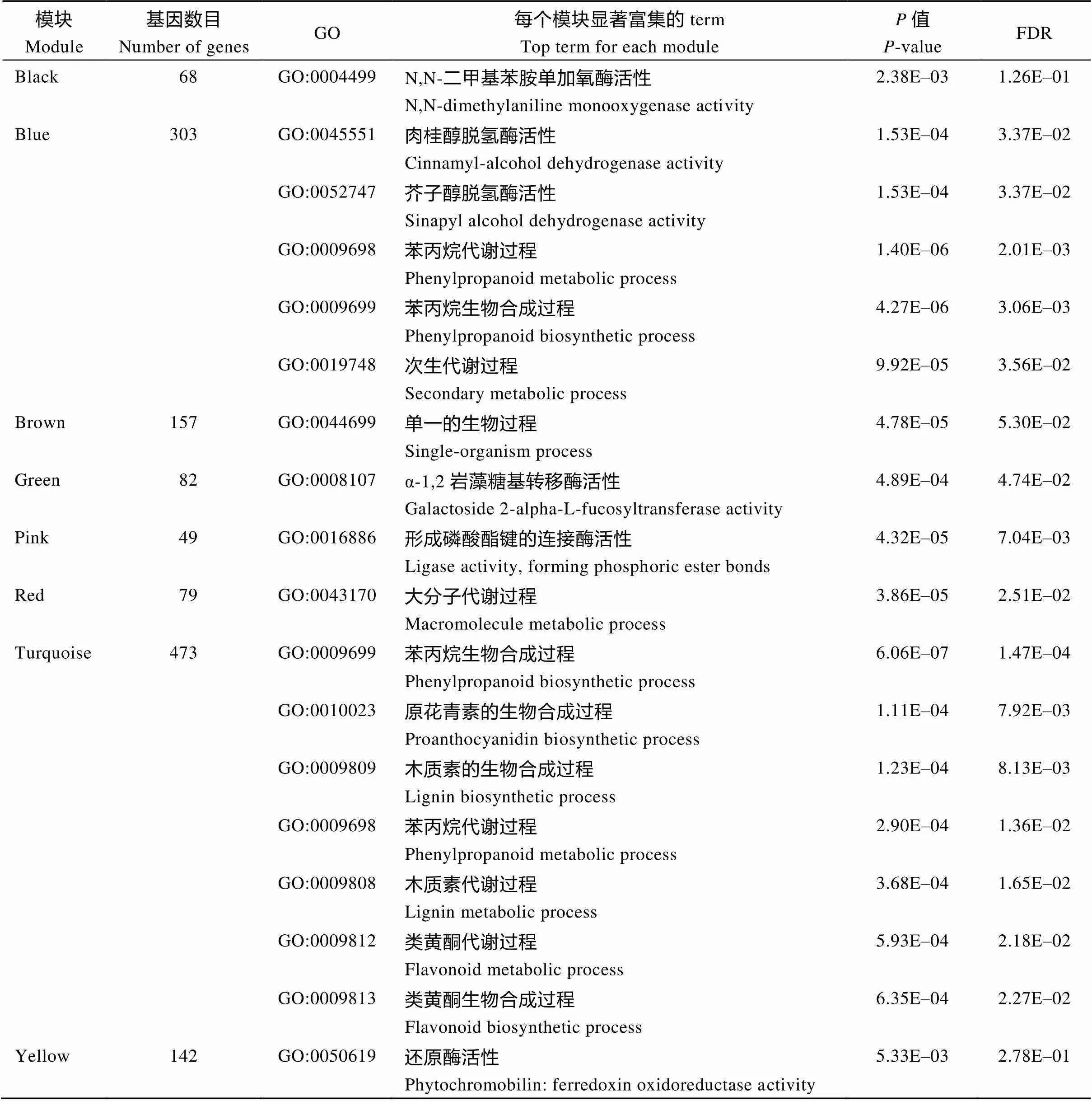
表1 各模块GO富集情况(部分)
2.3 黄籽相关模块的分析及枢纽基因(hub gene)的筛选
对turquoise模块进行KEGG富集分析, 最显著的代谢通路为类黄酮生物合成途径(ko00941,= 1.50E–07)和苯丙烷生物合成途径(ko00940,= 1.88E–04)。在这个模块中, 有7个基因属于类黄酮生物合成途径, 其中6个在模块中连通性排名前5%; 7个基因属于苯丙烷生物合成途径, 其中1个基因在这个模块中连通性排名前五。有4个基因在这个模块中连通性排名前10% (图3)。利用OS-tools网络图工具, 选择软阈值作为连通性计算方法, 对turquoise模块中weight ≥ 0.4的基因共表达网络图进行可视化,()、、()、()、()、()等基因处于该网络的中心, 具有较高的连通性。其中是苯丙烷代谢途径的关键基因,是已知的花色苷生物合成途径末端的一个关键酶基因, 而是类黄酮代谢途径的关键酶基因之一(图4)。作为原花青素途径上游代谢途径的关键基因, 它们之间分享大量相同的共表达基因, 表明在模块中, 基因功能之间存在显著的协同性。
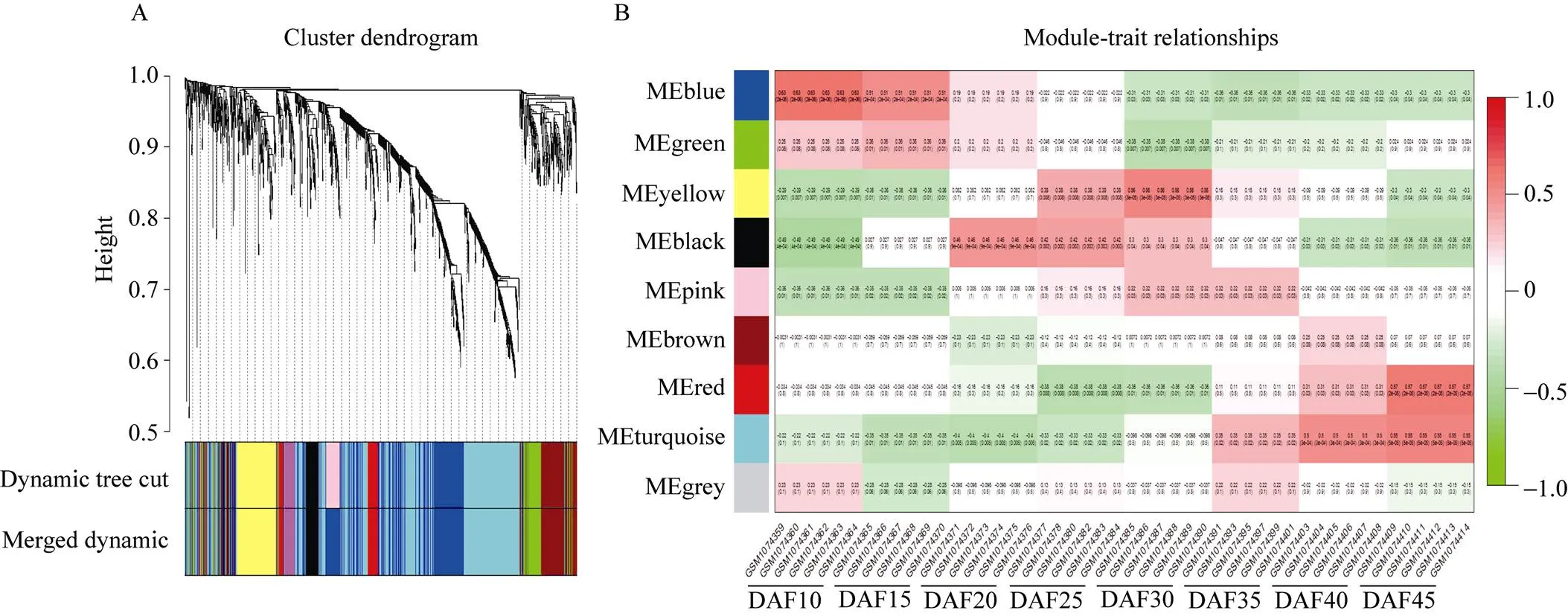
图2 基因共表达网络构建结果
A: 基因的聚类与模块构建; B: 特征与模块相关性分析。
A: the cluster of genes and construction of modules; B: correlation analysis between characteristics and modules.

图3 turquoise模块的KEGG富集分析
2.4 利用已知基因挖掘新的基因
基因是turquoise模块的一个核心基因, 其连通性在整个网络中排第九, 模块中排名第八, 在拟南芥中是花色苷生物合成途径末端的一个关键酶基因。此外,()和()基因虽然在模块内连通性排名仅为22和269, 但它们的拟南芥同源基因是原花青素途径的关键基因(编码花青素还原酶ANR是类黄酮途径中决定底物是否转向原花青素途径的关键基因[13];编码的LAC15参与原花青素的进一步多聚化和氧化, 赋予种皮棕色[14])。本研究分别以、和为中心, 筛选连通性排名前20的基因构建局部调控网络(图5-A)。整个局部网络共54个基因, 其中, 多个基因已知在苯丙烷合成代谢、类黄酮合成代谢中发挥着作用(表2)。

图4 turquoise模块内的基因共表达网络
()基因是blue模块的一个核心基因, 其连通性在模块中排名第3, 在拟南芥中对种皮色素的形成有重要作用。本研究以基因为核心, 筛选weight ≥ 0.3的调控关系以及相关基因构建局部调控网络(图5-B)。整个局部网络包含95个基因, 通过GO富集分析发现这个亚模块有8个term显著富集, 包括苯丙烷代谢过程(GO:0009698,= 1.41E–06)、次生代谢过程(GO:0019748,= 6.20E–06)、次生代谢产物生物合成过程(GO:0044550,= 1.52E–06)等。其中, 多个基因直接或间接参与苯丙烷-类黄酮-原花青素途径(表2)。这些信息在常规的GWAS分析中并没有被检测到, 只有通过WGCNA分析才被挖掘出来。
3 讨论
GWAS在动植物复杂性状研究上的应用, 很大程度上增强了研究者对这些复杂性状遗传机制的理解。但是, GWAS应用的难点在于有效过滤假阳性的同时挖掘微效作用位点。作为主要依赖统计分析的研究方法, GWAS在检测的过程中会引入大量的假阳性结果, 为了控制多重检验带来的虚报, 一般采用多重校正的方法[15]。然而, 无论采用何种校正方法, 都不能从根本上解决假阳性关联问题, 过于保守的校正还会导致假阴性结果的出现。此外, 微效作用位点对性状的贡献率都非常小, 经过多重校正后往往达不到显著水平, 其显著性淹没在背景噪音中, 难以被准确挖掘出来。为了解决这一难题, 多位学者提出了不同的策略, 如基于单体型的关联分析(Haplotype-based GWAS)[16]、基于基因的关联分析(Gene-based GWAS)[17]、利用CNV变异的GWAS[18]以及类似于Haplotype-based GWAS的RTM-GWAS方法[19]等。Farber[10]首次以WGCNA分析作为对GWAS分析的补充, 挖掘影响骨密度的微效作用位点, 其结果表明, 使用WGCNA策略对GWAS分析得到的大量的“名义”显著基因进行筛选, 能够提高二阶段关联分析验证的验证率。本研究整合GWAS和WGCNA, 挖掘影响黄籽表型相关的微效作用位点, 发现了大量与种皮色素形成相关的基因, 例如等原花青素途径的关键基因, 这些基因在我们前期的GWAS分析中因没有达到校正阈值而未被发现, 说明整合GWAS和WGCNA分析能够提高对黄籽表型微效作用位点的挖掘效率。

图5 枢纽基因相关的局部调控网络
红圈强调的基因参与类黄酮-原花青素途径。A: turquoise模块; B: blue模块。
The highlighted genes involved in flavonoid-proanthocyanidin pathways. A: turquoise module; B: blue module.
甘蓝型油菜种子发育过程中种皮色泽的变化主要受原花色素、多酚和木质素及其衍生物等重要次生代谢物质的影响[20], 控制类黄酮合成途径相关基因在种皮色泽形成中扮演着重要角色[21-25]。原花青素途径是类黄酮代谢途径中的重要分支之一, 同时在拟南芥中该途径的产物能够特异地在种皮中积累,是种皮颜色变化的主要原因[13]。在本研究中, 与黄籽表型相关性最高的turquoise模块显著地富集到了类黄酮合成代谢过程、原花青素的生物合成过程等GO term, 同时基于共表达网络hub gene挖掘出的基因大部分参与了苯丙烷-类黄酮途径, 而这些基因大部分未被传统的GWAS分析发现。此外, turquoise模块和blue模块均富集到了苯丙烷和木质素途径, 作为类黄酮途径的上游途径和相邻途径, 相关基因被划分到同一模块内, 表明其在转录水平上的相关性和功能上的协调性。上述结果进一步表明, 整合GWAS和WGCNA分析不仅可以提高对微效作用位点的挖掘效率, 而且也大大提升了GWAS的发展空间, 使其能够进一步为人类了解动植物复杂性状的遗传基础及分子机制提供更多的线索。
本研究使用相对宽松的FDR进行多重校正, 筛选出超过1800个名义上的关联基因, 但5%的FDR阈值线依然高达3.62 (= 2.41E–4, –lg () ≈ 3.62), 导致忽略掉了一部分更微效的位点。因此, 在以后的研究中, 整合GWAS和WGCNA分析必须协调好最大限度挖掘微效位点和尽可能过滤掉假阳性结果以避免消耗大量计算资源。此外, 在表型特异模块内基因的梳理过程中, 本研究发现原本显著关联的部分基因(和)并没有被划分入turquoise模块或者blue模块, 推测WGCNA共表达模块的划分是基于基因间表达量的相关性, 而基因的表达量受多种因素影响, 导致一部分真实关联基因被划分到其他模块。尽管如此, 整合GWAS和WGCNA分析得到的这些信息, 仍然可以为其他的实验提供一些参考, 同时也可为探究甘蓝型油菜黄籽基因资源信息、丰富分子育种理论奠定基础。
4 结论
采用整合全基因组关联分析(GWAS)和权重基因共表达网络分析(WGCNA)的策略, 挖掘油菜黄籽性状微效作用位点, 2年共检测到199个SNP位点, 在SNP位点附近共挖掘出1826个名义候选基因。基于候选基因表达谱数据构建了8个共表达模块, 其中turquoise模块和blue模块与黄籽表型相关性最高。、、等苯丙烷代谢途径、类黄酮途径的关键酶基因为turquoise模块的枢纽基因(hub gene)。通过已知的黄籽相关基因, 挖掘出了一部分黄籽微效作用基因, 这些基因多参与苯丙烷、类黄酮以及原花青素代谢途径。
[1] Wittkop B, Snowdon R J, Friedt W. Status and perspectives of breeding for enhanced yield and quality of oilseed crops for Europe., 2009, 170: 131
[2] 张永泰, 李爱民, 蒋金金, 王娟, 王幼平. 新型黄籽甘蓝型油菜的获得及其遗传规律分析. 中国油料作物学报, 2011, 33: 302–306 Zhang Y T, Li A M, Jiang J J, Wang J, Wang Y P. Discovery and genetics of new type of yellow seed., 2011, 33: 302–306 (in Chinese with English abstract)
[3] Remington D L, Thornsberry J M, Matsuoka Y, Wilson L M, Whitt S R, Doebley J, Kresovich S, Goodman M M, and Buckler E S. Structure of linkage disequilibrium and phenotypic associations in the maize genome., 2001, 98: 11479–11484
[4] Hasan M, Friedt W, Pons-Kuehnemann J, Freitag N M, Link K, Snowdon R J. Association of gene-linked SSR markers to seed glucosinolate content in oilseed rape (ssp).2008, 116: 1035–1049
[5] Wang J, Xian X H, Xu X F, Qu C M, Lu K, Li J N, Liu L Z. Genome wide association mapping of seed coat color in., 2017, 65: 5229–5237
[6] 杨小红, 严建兵, 郑艳萍, 余建明, 李建生. 植物数量性状关联分析研究进展, 作物学报, 2007, 33: 523–530 Yang X H, Yan J B, Zheng Y P, Yu J M, Li J S. Reviews of association analysis for quantitative traits in plants., 2007, 33: 523–530 (in Chinese with English abstract)
[7] Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis., 2005, 4: 17
[8] 刘伟, 李立, 叶桦, 屠伟. 权重基因共表达网络分析在生物医学中的应用. 生物工程学报, 2017, 33: 1791–1801 Liu W, Li L, Ye H, Tu W. Weighted gene co-expression network analysis in biomedicine research., 2017, 33: 1791–1801 (in Chinese with English abstract)
[9] Weiss J N, Karma A, MacLellan W R, Deng M, Rau C D, Rees C N, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z L, Wang Y B, Lusis A J. “Good enough solutions” and the genetics of complex diseases., 2012, 111: 493–504
[10] Farber C R. Systems-level analysis of genome-wide association data., 2013, 3: 119–129
[11] Huang D, Koh C, Feurtado J A, Tsang E W, Cutler A J. MicroRNAs and their putative targets inseed maturation., 2013, 14:140
[12] Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis., 2008, 9: 559
[13] 甘蓓, 杨红玉. 拟南芥中类黄酮代谢途径及其调控. 安徽农业科学, 2008, 36: 5290–5292 Gan B, Yang H Y. Metabolic approach of flavonoids and its regulation in., 2008, 36: 5290–5292 (in Chinese with English abstract)
[14] Pourcel L, Routaboul J M, Kerhoas L, Caboche M, Lepiniec L, Debeaujon I.encodes a laccase-like enzyme involved in oxidative polymerization of flavonoids inseed coat., 2005, 17: 2966–2980
[15] 黄杨岳, 孔祥祯, 甄宗雷, 刘嘉. 全基因组关联研究中的多重校正方法比较. 心理科学进展, 2013, 21: 1874–1882 Huang Y Y, Kong X Z, Zhen Z L, Liu J. The comparison of multiple testing corrections methods in genome-wide association studies., 2013, 21: 1874–1882
[16] Sato S, Uemoto Y, Kikuchi T, Egawa S, Kohira K, Saito S, Sakuma H, Miyashita S, Arata S, Kojima T, Suzuki K. SNP- and haplotype-based genome-wide association studies for growth, carcass, and meat quality traits in a Duroc multigenerational population., 2016, 17: 60
[17] Zhang W, Li J, Guo Y, Zhang L, Xu L, Gao X, Zhu B, Gao H, Ni H, Chen YMulti-strategy genome-wide association studies identify the DCAF16-NCAPG region as a susceptibility locus for average daily gain in cattle., 2016, 6: 38073
[18] Marshall C R, Howrigan D P, Merico D, Thiruvahindrapuram B, Wu W, Greer D S, Antaki D, Shetty A, Holmans P A, Pinto D, Gujral M, Brandler W M, Malhotra D, Wang Z, Fajarado K V F, Maile M S, Ripke S, Agartz I, Albus M, Alexander M, Amin F, Atkins J, Bacanu S A, Belliveau R A, Bergen S E, Bertalan M, Bevilacqua E, Bigdeli T B, Black D W, Bruggeman R, Buccola N G, Buckner R L, Bulik-Sullivan B, Byerley W, Cahn W, Cai G, Cairns M J, Campion D, Cantor R M, Carr V J, Carrera N, Catts S V, Chambert K D, Cheng W, Cloninger C R, Cohen D, Cormican P, Craddock N, Crespo-Facorro B, Crowley J J, Curtis D, Davidson M, Davis K L, Degenhardt F, Del Favero J, DeLisi L E, Dikeos D, Dinan T, Djurovic S, Donohoe G, Drapeau E, Duan J, Dudbridge F, Eichhammer P, Eriksson J, Escott-Price V, Essioux L, Fanous A H, Farh K H, Farrell M S, Frank J, Franke L, Freedman R, Freimer N B, Friedman J I, Forstner A J, Fromer M, Genovese G, Georgieva L, Gershon E S, Giegling I, Giusti-Rodríguez P, Godard S, Goldstein J I, Gratten J, de Haan L, Hamshere M L, Hansen M, Hansen T, Haroutunian V, Hartmann A M, Henskens F A, Herms S, Hirschhorn J N, Hoffmann P, Hofman A, Huang H, Ikeda M, Joa I, Kähler A K, Kahn R S, Kalaydjieva L, Karjalainen J, Kavanagh D, Keller M C, Kelly B J, Kennedy J L, Kim Y, Knowles J A, Konte B, Laurent C, Lee P, Lee S H, Legge S E, Lerer B, Levy D L, Liang K Y, Lieberman J, Lönnqvist J, Loughland C M, Magnusson P K E, Maher B S, Maier W, Mallet J, Mattheisen M, Mattingsdal M, McCarley R W, McDonald C, McIntosh A M, Meier S, Meijer C J, Melle I, Mesholam-Gately R I, Metspalu A, Michie P T, Milani L, Milanova V, Mokrab Y, Morris D W, Müller-Myhsok B, Murphy K C, Murray R M, Myin-Germeys I, Nenadic I, Nertney D A, Nestadt G, Nicodemus KK, Nisenbaum L, Nordin A, O’Callaghan E, O’Dushlaine C, Oh S Y, Olincy A, Olsen L, O'Neill F A, Van Os J, Pantelis C, Papadimitriou G N, Parkhomenko E, Pato M T, Paunio T, Perkins D O, Pers T H, Pietiläinen O, Pimm J, Pocklington A J, Powell J, Price A, Pulver A E, Purcell S M, Quested D, Rasmussen H B, Reichenberg A, Reimers M A, Richards A L, Roffman J L, Roussos P, Ruderfer D M, Salomaa V, Sanders A R, Savitz A, Schall U, Schulze T G, Schwab S G, Scolnick E M, Scott R J, Seidman L J, Shi J, Silverman J M, Smoller J W, Söderman E, Spencer C C A, Stahl E A, Strengman E, Strohmaier J, Stroup T S, Suvisaari J, Svrakic D M, Szatkiewicz J P, Thirumalai S, Tooney P A, Veijola J, Visscher P M, Waddington J, Walsh D, Webb B T, Weiser M, Wildenauer D B, Williams N M, Williams S, Witt S H, Wolen A R, Wormley B K, Wray N R, Wu J Q, Zai C C, Adolfsson R, Andreassen O A, Blackwood D H R, Bramon E, Buxbaum J D, Cichon S, Collier D A, Corvin A, Daly M J, Darvasi A, Domenici E, Esko T, Gejman P V, Gill M, Gurling H, Hultman C M, Iwata N, Jablensky A V, Jönsson E G, Kendler K S, Kirov G, Knight J, Levinson D F, Li Q S, McCarroll S A, McQuillin A, Moran J L, Mowry B J, Nöthen M M, Ophoff R A, Owen M J, Palotie A, Pato C N, Petryshen T L, Posthuma D, Rietschel M, Riley B P, Rujescu D, Sklar P, St Clair D, Walters J T R, Werge T, Sullivan P F, O’Donovan M C, Scherer S W, Neale B M, Sebat J. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects., 2017, 49: 27–35
[19] He J, Meng S, Zhao T, Xing G, Yang S, Li Y, Guan R, Lu J, Wang Y, Xia Q, Yang B, Gai J. An innovative procedure of genome–wide association analysis fits studies on germplasm population and plant breeding., 2017, 130: 2327–2343
[20] 叶小利, 李加纳, 唐章林, 梁颖, 谌利. 甘蓝型油菜种皮色泽及相关性状的研究. 作物学报, 2001, 27: 550–556 Ye X L, Li J N, Tang Z L, Liang Y, Chen L. Study on seed coat color and related characters of., 2001, 27: 550–556 (in Chinese with English abstract)
[21] Fu F Y, Liu L Z, Chai Y R, Chen L, Yang T, Jin M Y, Ma A F, Yan X Y, Zhang Z S, Li J N. Localization of QTLs for seed color using recombinant inbred lines ofin different environments., 2007, 50: 840–854
[22] Chai Y R, Lei B, Huang H L, Li J N, Yin J M, Tang Z L, Wang R, Chen L.genes fromand parental species: cloning, evolution, and differential involvement in yellow seed trait., 2009, 281: 109–123
[23] Zhang J, Lu Y, Yuan Y, Zhang X, Geng J, Chen Y, Cloutier S, McVetty P B, Li G. Map-based cloning and characterization of a gene controlling hairiness and seed coat color traits in., 2009, 69: 553–563
[24] Stein A, Wittkop B, Liu L, Obermeier C, Friedt W, Snowdon R J. Dissection of a major QTL for seed colour and fibre content inreveals colocalization with candidate genes for phenylpropanoid biosynthesis and flavonoid deposition., 2013, 132: 382–389
[25] Padmaja L K, Agarwal P, Gupta V, Mukhopadhyay A, Sodhi Y S, Pental D, Pradhan A K. Natural mutations in two homoeologousgenes control yellow seed coat trait in allotetraploid(AABB)., 2014, 127: 339–347
Mining Yellow-seeded Micro Effect Loci inby Integrated GWAS and WGCNA Analysis
XIAN Xiao-Hua1,**, WANG Jia1,2,**, XU Xin-Fu1, QU Cun-Min1, LU Kun1, LI Jia-Na1, and LIU Lie-Zhao1,*
1College of Agronomy and Biotechnology / Academy of Agricultural Sciences, Southwest University, Chongqing 400715, China;2Nanchong Academy of Agricultural Sciences, Nanchong 637000, Sichuan, China
is one of the most important oil crops in the world, and developing yellow-seededwith improved qualities is a major breeding goal. The yellow-seeded minor genes were mined by genome-wide association study (GWAS) and weighted gene co-expression network analysis (WGCNA) with 520 representative varieties (or lines) and the transcriptional data at eight time points during the seed development. The 199 SNPs and 1826 nominally significant GWAS candidate genes were detected. Weighted gene co-expression network analysis was performed using the WGCNA R package to construct the resulting network composing eight distinct gene modules. Among them, the turquoise module and the blue module were related to the seed coat color based on gene function enrichment analysis.,, and, the key enzymes genes of phenylpropane metabolic pathway and flavonoid metabolic pathway were found in turquoise module. Through the characterization of module content and topology, we mined a number of micro effect genes based on known yellow-seed related genes mainly involved in the phenylpropanoid metabolic process, flavonoid metabolic process and proanthocyanidin biosynthetic process. This information of minor loci and candidate genes should be useful in the breeding for yellow-seeded.
; yellow-seeded; GWAS; WGCNA
2018-06-09;
2018-06-11.
10.3724/SP.J.1006.2018.01105
刘列钊, E-mail: liezhao2003@126.com, Tel: 023-68251383
**同等贡献(Contributed equally to this work)
鲜小华, E-mail: xxm920423@126.com, 王嘉, E-mail: wangjia0724@126.com
2018-01-29;
本研究由国家自然科学基金项目(31771830), 重庆市科委项目(cstc2016shmszx80083)和中央高校基本科研业务费专项(XDJK2017A009)项目资助。
This study was supported by the National Natural Science Foundation of China (31771830), the Science and Technology Committee of Chongqing (cstc2016shmszx80083), and the Fundamental Research Funds for the Central Universities (XDJK2017A009).
URL:http://kns.cnki.net/kcms/detail/11.1809.S.20180611.0904.008.html
