解偶联蛋白2过表达对脓毒症大鼠心肌细胞线粒体的保护作用
2018-07-27郑贵浪郭予雄吕娟娟黄锦达曾其毅
郑贵浪,郭予雄,吕娟娟,黄锦达,刘 翠,曾其毅
(1.广东省人民医院 广东省医学科学院儿科,广东 广州 510030;2.南方医科大学珠江医院儿科,广东 广州 510220)
脓毒症是由感染诱发的机体调节失衡所致的危及生命的器官功能障碍[1],其高发病率和高死亡率耗费了大量的医疗资源。研究[2]显示:脓毒症时心功能障碍的发生率高达40%,且与其导致的线粒体功能障碍有关。解偶联蛋白2 ( uncoupling protein 2 ,UCP2)属于线粒体阴离子通道家族成员之一,广泛分布于心、肺和脑等器官和组织中,在多种病理生理过程中发挥重要作用。研究[3]表明:UCP2参与动脉粥样硬化的形成,参与免疫应答反应、食物的摄入和多种代谢性疾病的病理生理过程。体内外研究[3-4]显示:UCP2上调可以通过调控膜电位进而保护神经元,而UCP2基因敲除则会导致线粒体活性氧(ROS)产生明显增多以致损害神经元。近年来研究[5]显示:UCP2可作为临床严重脓毒症的特异性标记物。对于心肌细胞,尤其在脓毒症状态下的心肌细胞中UCP2的作用,近年来报道较少。本课题组前期研究[6]显示:通过干扰心肌中UCP2的表达,脓毒症可使心肌细胞线粒体结构及功能明显受损。为进一步揭示UCP2的作用,本研究在构建UCP2过表达的稳转株心肌细胞基础上,采用脂多糖/聚糖肽(LPS/PepG)刺激心肌细胞,通过检测线粒体形态与功能的相关指标,揭示UCP2在脓毒症心肌细胞中的保护作用及其可能机制。
1 材料与方法
1.1 UCP2过表达H9C2心肌细胞稳定株的构建提取大鼠总RNA,采用常规基因操作技术,将UCP2基因构建到pMD19-T质粒;以EcoRⅠ和NotⅠ为插入位点,将UCP2基因克隆到慢病毒载体(PHBLV-IRES-ZsGreen-PGK-puro穿梭载体),通过慢病毒包装及滴度测定,将克隆成功的UCP2慢病毒穿梭载体感染H9C2心肌细胞,筛选出UCP2过表达H9C2细胞株进行实验。
1.2 细胞培养和分组将H9C2心肌细胞随机分成4组:①正常对照组,给予等量生理盐水刺激;② LPS/PepG 刺激组,同时给予LPS和PepG刺激;③ 空病毒组,空病毒载体(PHBLV)转染H9C2细胞,同时给予LPS和PepG刺激;④ 过表达组,UCP2过表达H9C2细胞同时给予LPS和PepG刺激。上述LPS终浓度为2 mg·L-1,PepG终浓度为 20 mg·L-1。
1.3 RT-PCR和Western blotting法检测心肌细胞中UCP2 mRNA和蛋白表达水平① RT-PCR法。UCP2引物:GGGCACCTGTGGTGCTACCTG(R),ATGAGCTTTGCCTCCGTCCGC(F),扩增片段长度为118 bp;GAPDH引物:GAAGATGGTGATGGGTTTCC(R),CTACAACGACCCCTTCA-TTG(F),扩增片段长度为136 bp;18sRNA引物:CCATCCAATCGG TAGTAGC(R),GTAATGGCGG GTCATAAG(F),扩增片段长度为80 bp。TRIzol法提取细胞总RNA,逆转录反应,实时荧光定量PCR扩增目的基因。根据实时荧光定量PCR仪检测到的SYBR Green Ⅱ的荧光信号,计算目的基因UCP2的相对表达水平,即目的基因的表达水平相对于对照组的变化倍数。② Western blotting法。裂解液裂解H9C2细胞,离心后取上清,提取总蛋白并测定蛋白浓度;根据BCA蛋白定量试剂盒进行蛋白定量;提取总蛋白,加入SDS蛋白上样缓冲液煮沸保存备用;Western blotting 法检测蛋白表达,采用化学发光法(ECL)进行显影。利用Kodak In-Vivo Imagine System F化学发光/活体成像分析系统采集图像,Gel-Pro analyzer 4.0软件比较各条带的灰度值,计算目的蛋白的表达水平。
1.4 心肌细胞中肌酸激酶(CK)和肿瘤坏死因子α(TNF-α)水平检测采用分光光度计法检测心肌细胞中CK水平,采用ELISA法检测心肌细胞中TNF-α水平,具体操作步骤按试剂盒说明书进行(CK测定试剂盒购自南京建成生物工程研究所,ELISA试剂盒购自北京博凌科为生物科技有限公司)。CK水平升高提示心肌细胞损害,TNF-α水平升高提示炎症信号通路启动。
1.5 电镜观察线粒体超微结构常规制备培养的H9C2心肌细胞的电镜标本,超薄切片染色后电镜观察H9C2细胞线粒体的超微结构。
1.6 流式细胞术和激光共聚焦显微镜检测线粒体膜电位(MMP)水平采用一种阳离子脂质荧光染料JC-1作为检测MMP指示剂。JC-1在低浓度时可发射绿色荧光,高浓度时发射光为红色荧光。MMP升高时,JC-1聚合体形式增加,FL-2/FL-1比值升高;MMP下降时,JC-1单体形式增加,FL-2/FL-1比值降低。利用这一特性,本实验通过流式细胞术及激光共聚焦技术检测各组心肌细胞中MMP水平,同时设阳性对照组。具体操作步骤按照细胞线粒体分离试剂盒(南京建成生物科技有限公司,货号G003)和MMP检测试剂盒(凯基生物科技发展有限公司,货号KGA604)说明书进行。
1.7 多功能酶标仪检测心肌细胞中线粒体ROS和三磷酸腺苷(ATP)水平采用多功能酶标仪检测心肌细胞中线粒体 ROS和ATP水平。具体操作步骤按试剂盒说明书进行。ROS检测试剂盒(货号S0033)和ATP检测试剂盒(货号A016-1)均购自碧云天生物技术研究所。

2 结 果
2.1 各组H9C2心肌细胞中UCP2 mRNA和蛋白表达水平与正常对照组比较,LPS/PepG 刺激组 H9C2心肌细胞中UCP2 mRNA和蛋白表达水平明显升高(P< 0.05);过表达组H9C2心肌细胞中UCP2 mRNA和蛋白表达水平均明显高于正常对照组、LPS/PepG 刺激组和空病毒组(P< 0.05);与LPS/PepG 刺激组比较,空病毒组H9C2心肌细胞中UCP2 mRNA和蛋白表达水平差异无统计学意义(P>0.05)。上述结果提示H9C2心肌细胞UCP2过表达模型构建成功。见图1和表1。
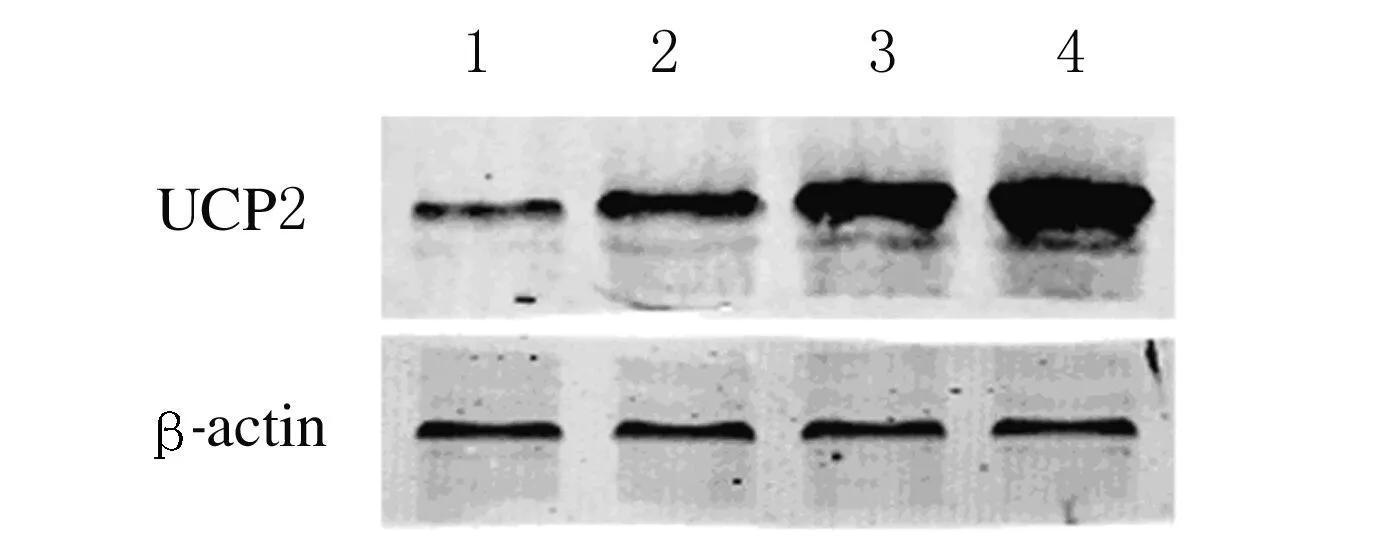
Lane 1:Normal control group;Lane 2:LPS/PePG group;Lane 3:PHBLV group;Lane 4:PHBLV-UCP2 group.
图1 各组H9C2心肌细胞中UCP2蛋白表达电泳图
Fig.1 Electrophoregram of UCP2 protein in H9C2 cells in various groups
2.2 各组H9C2心肌细胞中CK和TNF-α水平 与正常对照组比较, LPS/PepG 刺激组H9C2心肌细胞中CK和TNF-α水平均明显升高(P<0.05);与LPS/PepG 刺激组和空病毒组比较,过表达组H9C2细胞中CK和TNF-α水平均明显降低(P<0.05)。空病毒组H9C2心肌细胞中CK和TNF-α水平与LPS/PepG 刺激组比较差异无统计学意义(P>0.05)。见表1。
2.3 各组H9C2心肌细胞线粒体超微结构 电镜下观察:正常对照组心肌细胞线粒体边界清楚,基质均匀,线粒体嵴致密,形态正常。LPS/PepG 刺激组心肌细胞线粒体边界模糊不清,体积肿胀明显,基质密度降低,部分线粒体内膜破裂、嵴疏松溶解,甚至出现嵴断裂现象,可见空泡样变。与LPS/PepG 刺激组比较,过表达组心肌细胞线粒体形态有所改善,表现为线粒体肿胀减轻,基质密度增高,内膜破裂及嵴断裂减少,空泡样变不明显,但尚未能完全恢复到生理盐水组的水平。空病毒组心肌细胞线粒体电镜下结构改变与LPS/PepG 刺激组相似。见图2。
表1 各组H9C2心肌细胞中UCP2 mRNA和蛋白表达水平及CK、TNF-α水平
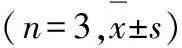

GroupUCP2 mRNAUCP2 proteinCK [λB/(U·L-1)]TNF-α[ρB/(ng·L-1)]Normal control 1.05±0.050.25±0.023.17±0.054.23±0.13LPS/PepG 1.43±0.02∗0.35±0.01∗6.45±0.07∗6.69±0.09∗PHBLV 1.47±0.02∗0.35±0.02∗6.40±0.076.73±0.13∗PHBLV-UCP2 1.94±0.01∗△# 0.57±0.01∗△#3.43±0.04△#5.30±0.27△#F466.571.81 328.9837.3P0.000.000.000.00
*P< 0.05 compared with normal control group;△P< 0.05 compared with LPS/PepG group;#P< 0.05 compared with PHBLV group.
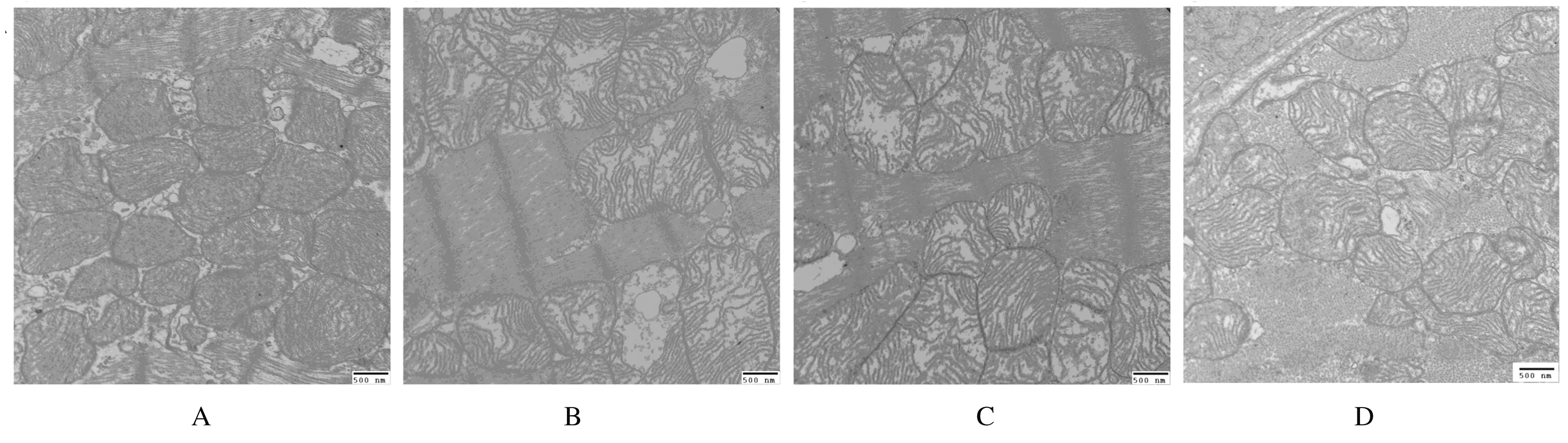
A:Normal control group;B: LPS/PepG group;C: PHBLV group;D: PHBLV-UCP2 group.
2.4 各组H9C2心肌细胞中MMP水平 激光共聚焦显微镜定性观察:与正常对照组比较,LPS/PepG刺激组、空病毒组和过表达组心肌细胞中红色荧光(JC-1聚合体)/绿色荧光(JC-1单体)明显下降;与LPS/PepG 刺激组和空病毒组比较,过表达组心肌细胞中红色荧光(JC-1聚合体)/绿色荧光(JC-1单体)明显上升。阳性对照组几乎不见红色荧光。见图3(插页一)。与正常对照组比较,LPS/PepG 刺激组和空病毒组心肌细胞中MMP水平明显降低(P< 0.05);过表达组心肌细胞中MMP水平明显高于LPS/PepG 刺激组和空病毒组(P< 0.05),但仍然低于正常对照组(P< 0.05);与LPS/PepG 刺激组比较,空病毒组心肌细胞MMP水平差异无统计学意义(P>0.05)。见表2。
2.5 各组H9C2心肌细胞中线粒体ROS和ATP水平 与正常对照组比较,LPS/PepG 刺激组和空病毒组心肌细胞中线粒体ROS水平明显升高(P<0.05),ATP水平明显降低(P<0.05);与LPS/PepG 刺激组和空病毒组比较,过表达组心肌细胞中线粒体ROS水平明显减低(P<0.05),ATP水平明显升高(P<0.05)。与LPS/PepG组比较,空病毒组心肌细胞线粒体ROS和ATP水平差异无统计学意义(P>0.05)。见表2。
表2 各组H9C2心肌细胞中MMP和线粒体ROS及ATP水平

GroupMMPROSATP[mB/(μmol·g-1)]Normal control 0.424±0.0061.00±0.146.19±0.07LPS/PepG 0.213±0.005∗2.53±0.03∗2.65±0.05∗PHBLV0.206±0.006∗2.49±0.04∗2.55±0.12∗PHBLV-UCP2 0.399±0.007∗△#1.73±0.06△# 4.54±0.08△#F567.8183.81 322.6P0.000.000.00
*P< 0.05 compared with normal control group;△P< 0.05 compared with LPS/PepG group;#P< 0.05 compared with PHBLV group.
3 讨 论
脓毒症是感染诱导机体调节失衡所致的危及生命的器官功能不全,严重脓毒症是指脓毒症并发器官功能不全或组织低灌注。Hartman等[7]回顾性研究发现:2005年新生儿严重脓毒症的发病率较1995年升高了1倍。脓毒症诱导的心肌功能损害(SIMD)是严重脓毒症的常见并发症,其机制十分复杂[8]。
脓毒症的动物模型比较成熟,细胞模型目前缺乏统一的标准[9-11]。本研究采用来自金黄色葡萄球菌细胞壁上的PepG与来自革兰阴性菌LPS组成混合剂,刺激H9C2心肌细胞后,检测到培养液中的CK及TNF-α水平明显升高,提示脓毒症的细胞模型构建成功;LPS/PepG刺激H9C2心肌细胞后,CK及TNF-α水平升高,提示LPS/PepG损害了心肌细胞,同时启动了炎症通路;增加UCP2在心肌细胞中的表达后,CK及TNF-α水平明显降低,提示UCP2对脓毒症心肌线粒体起保护作用,其具体机制可能与UCP2调节MMP有关。
心肌细胞富含线粒体,可提供大量ATP,同时也是大量产生ROS的场所。本研究显示:UCP2过表达可以抑制心肌线粒体ROS的过多生成,这可能与MMP趋向正常状态有关。Kizaki等[12]和Blanc等[13]通过巨噬细胞转染UCP2 cDNA后发现:巨噬细胞产生的ROS量减少,而血细胞中缺乏UCP2会导致ROS合成增多,导致动脉粥样硬化的恶化,与本实验结果相似。Nègre-Salvayre等[14]研究发现:在肝、脾及胸腺中,GDP(UCP2解偶联特效抑制剂)可以升高膜电位并引起ROS产生增多。上述实验结果提示:UCP2可以调控ROS的合成,但其具体机制目前尚不完全清楚。
线粒体ATP水平是线粒体功能的重要指标。本研究结果显示:脓毒症时UCP2表达被抑制,MMP破坏,O2利用障碍,导致ATP合成明显减少。通过慢病毒介导UCP2过表达后,线粒体ATP水平明显回升,趋近正常水平,提示UCP2对于维持线粒体ATP水平有益。可能与UCP2通过解偶联作用减少ROS合成有关。但UCP2与线粒体ATP合成之间的关系,目前争论较多[15-16],实验结果也不尽相同[17],其机制可能与线粒体的结构有关。
本实验电镜检测结果显示:LPS/PepG混合剂可导致心肌细胞线粒体形态结构破坏,表现为线粒体的边界模糊不清,体积明显肿胀,基质密度降低,部分线粒体内膜破裂、嵴疏松溶解,甚至出现嵴断裂现象,可见空泡样变。Welty-Wolf等[18]在狒狒脓毒症模型中发现:注入大肠埃希菌后12 h,狒狒的骨骼肌线粒体就出现线粒体嵴模糊,基质空泡化和内膜断裂,提示脓毒症导致线粒体结构的改变。Vanhorebeek等[19]在探讨胰岛素对脓毒症患者线粒体的保护中发现:死于脓毒症的20例ICU患者肝脏和肌肉组织的线粒体均明显的肿胀、基质密度降低,部分线粒体内膜破裂,并且线粒体结构的损害与呼吸链复合体功能有关联。此外,在实验动物[20]和脓毒症患者[21]心肌细胞中也有类似发现。本研究通过慢病毒促使UCP2过表达后,线粒体的形态结构明显改善,其机制可能与ROS及MMP有关。
UCPs可降低线粒体外膜的质子势能,从而稳定MMP。UCP2可以通过解耦联作用,开放离子通道,H+不通过ATP合成酶直接从线粒体内膜渗漏到线粒体基质中,从而起到调控膜电位的作用,同时不合成ATP。本研究结果显示: 脓毒症导致心肌细胞MMP明显下降,与本课题组前期研究结果一致[6];UCP2过表达后,MMP接近正常水平,表明UCP2可促进脓毒症导致的MMP破坏的恢复,与传统的UCP2解偶联作用不一致,其中准确机制尚不明确。综合前期研究及其他学者[22-23]的研究结果,本文作者认为:UCP2通过解偶联作用调控MMP。此外,线粒体的形态结果和MMP也可能与线粒体内钙浓度及线粒体通透性膜转换孔的状态[24]等有关,但本实验未对上述指标进行检测。
综上所述,UCP2过表达可以抑制脓毒症所导致的大鼠心肌细胞的损害及炎症因子的释放,同时脓毒症所致的线粒体形态及功能的损害,可被UCP2过表达所逆转或减轻。UCP2过表达对心肌细胞线粒体的保护作用机制可能是UCP2通过解偶联作用调控心肌细胞MMP,减少ROS过多生成,维持线粒体的ATP合成,从而起到保护线粒体形态结构及功能的作用。
