嘧啶结构骨架类抗疟药研究进展
2018-06-12丁奇峰贺常虹谈嘉怡岳慧慧
丁奇峰,贺常虹,谈嘉怡,岳慧慧,薛 蔹,于 杨,黄 和,黄 菲
(南京工业大学 药学院,江苏 南京 211800)
疟疾是一种由疟原虫感染而引起的,能够危及人类生命的疾病。如今每年由疟原虫感染的病例达到2亿之多,并可造成超过438 000人死亡,且疟疾在非洲尤为严重,是非洲儿童和妇女主要的死亡原因之一[1]。现如今随着科技以及医疗卫生事业的发展,疟疾发病率以及致死率都在逐年递减,然而对恶性疟原虫却一直找不到有效的解决办法。恶性疟原虫已经对多种抗疟药(氯喹、奎宁等)产生了耐药性,其发病凶险,愈后差,致死率高达50%[2],严重威胁着人类生命健康,阻碍社会的发展。
疟原虫生命周期复杂,总的来看疟原虫的生命周期主要包括4个阶段:蚊虫传播阶段、肝脏阶段、血液阶段以及性成熟阶段(图1[3])。蚊虫叮咬将唾液中的疟原虫引入人体血液中,此后疟原虫到达肝脏和血液以逐渐成熟和繁殖。疟原虫在不同阶段都可以产生不同的表面抗原以逃避宿主的免疫清除,因此针对其每个阶段都必须考虑相应对的办法,以根除这种疾病。常见可寄生于人类的疟原虫有4种,即:间日疟原虫、卵形疟原虫、三日疟原虫和恶性疟原虫,它们分别可引起间日疟疾、卵形疟疾、三日疟疾和恶性疟疾。前3种疟疾目前都能得到有效的治疗,而恶性疟疾由于其发病的凶险性则一直是医学界难以攻破的课题。
鉴于全球恶性疟原虫已经对传统抗疟药氯喹产生了耐药性,在疟疾流行地区,媒介控制以及化学治疗是目前控制恶性疟疾的主要对策。媒介控制通过使用杀虫剂处理过的蚊帐以及清除生活区的停滞水等方法[4];化学治疗包括杀灭无性寄生虫以及对宿主采用支持疗法以增强其免疫力等方法来扼制恶性疟疾的蔓延。然而,疟疾现今的发病形势依旧非常严峻,青蒿素及其衍生物虽然具有高效和低毒的特点,并可有效对抗恶性疟原虫,但是在东南亚地区已经出现了恶性疟原虫青蒿素耐药性的案例[5-7]。因此,针对其耐药性,开发出更具抗性特点的新型抗疟药已迫在眉睫。

图2 苯并嘧啶类化合物结构Fig.2 Benzopyrimidine compounds structure diagram
嘧啶类抗疟药大多作用于疟原虫的二氢叶酸还原酶(DHFR),实现对该酶活性的抑制,从而阻断疟原虫四氢叶酸的合成途径,并由此发挥抗疟活性。按化学结构的不同,将近10年来这类化合物在抗疟活性及其构效关系(SAR)等方面的研究进展进行综述。

图1 疟原虫的传播阶段示意图Fig.1 Schematic diagram for parasite transmission stage
1 苯并嘧啶类化合物
在20世纪60年代就有报道称苯并嘧啶类化合物具有重要的抗疟活性[8],其嘧啶环上通常具有2个胺基,苯环上可引入各种不同的取代基。因此可以在苯并嘧啶骨架上引入不同的基团,以寻找可以克服耐药性的新化合物。
Paul等[8]通过高通量筛选,得到一系列苯并嘧啶类化合物,并对其抗疟活性进行了初步的分析。化合物1为筛选得出的代表化合物(图2)。经活性测试,化合物1对氯喹敏感的恶性疟原虫(3D7)的半抑制浓度(IC50)为0.106 μmol/L,对氯喹耐受的恶性疟原虫(W2)IC50为0.715 μmol/L,这说明该化合物具有很强的抗疟潜质。在这类化合物中,哌啶环上的取代基对抗疟活性的影响特别重要。化合物1中哌啶环对位上的甲基若调到邻位上形成化合物2,则其活性大大降低(对3D7的IC50为0.205 μmol/L,对W2的IC50大于1.25 μmol/L)。另外,Plouffe等[8]还进行了构效关系的探究,如图3所示:中间的骨架结构代表这类抗疟化合物,右上角的骨架代表还原型辅酶——NADPH,其余螺旋条带结构代表恶性疟原虫的二氢叶酸还原酶(DHFR),从图3可以看出:这类化合物与DHFR受体活性中心结合效果较好,因此具有一定的抗疟活性。

图3 化合物1、2的构效关系Fig.3 Structure-activity relationship of compounds 1,2
Malmquist等[9]从已知的组蛋白甲基转移酶抑制剂-BIX-01294(化合物3)出发,改造得到TM2-115(化合物4)。这2种化合物不同于传统的嘧啶类化合物作用机制,它们能抑制组蛋白甲基转移酶,使甲基不能转移到特定氨基酸的位置上[10],从而影响组蛋白正常的功能。鉴于组蛋白的赖氨酸甲基化过程在疟原虫的分化和增殖过程中至关重要,因此这类化合物可能具有的一个最大优势是对疟原虫生命周期的所有阶段都具有抑制作用。此外,化合物3可以用来参与干细胞的调制[11-12],在提高药物效力、选择性和细胞通透性方面也具有很好的潜力[13-18]。在抗疟活性方面,化合物3对W2(耐氯喹)、7G8(耐氯喹和乙胺嘧啶)和Dd2(耐氯喹、甲氟喹和乙胺嘧啶)这3种耐药性疟原虫均具有良好的抑制作用(IC50分别为52、40 和72 nmol/L),其衍生化合物4也具有相似活性。
Paul等[4]通过高通量筛选得到化合物5,并基于化合物5的结构进行多方位的改造,得到56个化合物,并具体分析了其构效关系及抗疟活性。如R1位置上单取代胺基基团(如5(a)和5(b))对抗疟活性是至关重要的(图2)。另外,甲氧基在苯环上的不同取代位点对活性也有较大影响。当甲氧基在R3及R4位置上时,其具有较好活性,半最大效应浓度(EC50)分别为124 和230 nmol/L;而当甲氧基在R2位置上时,其EC50为526 nmol/L,活性大大减弱。此外苯环上双取代甲氧基也不利于其抗疟活性。当在R4位置上引入卤素(Br、Cl和F)时,活性有所提升(EC50分别为125、113 和112 nmol/L),值得一提的是其选择性同时也得到很大程度的提高,选择指数(SI)分别为153、71和43。Paul等[4]还将R1位置引入苄胺,结果发现:整体活性仍有所保留,这样可以在R4位置上引入所需基团来改善化合物整体的物理化学性能。例如,当R1为苄胺,R4位置为甲氧基时,该化合物的疏水常数有所提高,水溶性也得到有效改善。为进一步改善疏水常数、溶解度以及选择性,Chang等[17]将与嘧啶合并的苯环改成吡啶环,R1位置为苄胺,R4位置为F,得到性能更优良的结构,其SI为320,疏水常数为4.6。在对构效关系和理化性质进行探索之后,综合测试了各类化合物的抗疟活性,发现化合物6拥有更全面的良好性能,其对恶性疟原虫的EC50为26 nmol/L,SI为240,疏水常数为4.2。
2 胺基嘧啶类化合物
2.1 二胺基嘧啶类化合物
二胺基嘧啶类化合物指的是在嘧啶环的2个位置上含有胺基基团的化合物。Yuthavong等[19]针对疟原虫二氢叶酸还原酶发生突变而产生耐药性的情况进行了深入的研究,化合物7~9是其研究的主要化合物(图4)。该团队初步研究就发现了化合物7对突变型的疟原虫二氢叶酸还原酶具有较高活性(IC50=18 nmol/L),然而小鼠口服的生物利用度却很差。因此,对化合物7进行改造得到化合物8和9。对比这2个化合物发现:化合物9对突变型的疟原虫活性较好(IC50=56 nmol/L),且生物利用度大大提高。此外,他们还对化合物9与二氢叶酸还原酶的结合方式进行了研究,通过蛋白质晶体衍射的手段得到图4(b)所示的构效关系图。从图4(b)可以看出:化合物9与疟原虫突变的酶有着部分的连接,而与人类的酶则无此类连接,说明化合物9具有一定的选择性。总的来说,化合物9对疟原虫的二氢叶酸还原酶的抑制遵循紧密结合-缓释动力学,其中抑制剂在酶的紧密结合构象状态上具有长的停留时间。这项研究还表明:化合物9不仅具有对野生型和耐药型疟疾有效的体外和体内抗疟活性,而且还有合适的药理学、药物代谢和安全性特征,因此可被进一步临床开发使用。
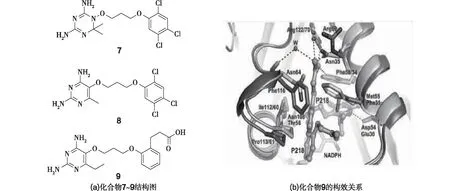
图4 化合物7~9结构图(a),化合物9的构效关系(b)Fig.4 Structure diagram for compounds 7-9 (a), structure-activity relationship of compound 9 (b)

图5 三胺基取代嘧啶类化合物构效关系Fig.5 Structure-activity relationship diagram of triamino substituted pyrimidines
2.2 三胺基嘧啶类化合物
三胺基嘧啶类化合物是指在嘧啶环的2、4和5位置上含有胺基基团的化合物,其各个胺基上又可被不同基团所取代,因此该类化合物种类多样,药效活性也各有差异。Srivastava等[20]经过筛选得到一系列三胺基嘧啶类化合物,并发现这类化合物拥有较长的半衰期,对恶性疟原虫效果较好,且在体外情况下不易自身产生耐药性。他们对此系列化合物构效关系进行了具体分析,结构与相关数据见图5。化合物10为其经高通量筛选所得的目标化合物,其抗疟活性、溶解度(solubility)以及对心脏离子通道(hERG)毒性都有待优化。经苯环上烷基取代的转变,得到化合物11,发现部分参数得到优化但其抗疟活性则大大降低,因此须进行进一步的改造。对化合物12来说,吡唑以及乙基的引入使对乙酰胆碱酯酶(AChE)的抑制作用增强,抗疟活性也有所增加。再次改造得到化合物13,其在C5上取代有N-甲基哌嗪,并且还引入了环丙基,具有最优的抗恶性疟原虫活性(IC50=9 nmol/L)、极好的溶解性(solubility>1 000 μmol/L),大鼠生物利用度(Rat F为80%)与化合物12相比也有很大提高。此外,化合物13对心脏离子通道以及乙酰胆碱酯酶抑制作用较弱(hERG IC50=33 μmol/L,AChE IC50>75 μmol/L),因此毒性较弱。综合分析,化合物13显示了良好的体内安全性以及与其他药物联合使用的抗疟潜力,因此可成为被临床测试的候选药物。

图7 噻吩并嘧啶类化合物结构Fig.7 The structure of thienopyrimidine compounds
3 喹啉并嘧啶类化合物
喹啉类骨架化合物曾被广泛应用于治疗疟疾,而嘧啶类骨架化合物则具有多种生物活性,例如:杀菌[21]、除草[22]、镇痛[23]、抗炎[24]和抗肿瘤[25]。Galmarini等[25]通过二胺基烷烃将喹啉与嘧啶相连,使得该分子具有足够的灵活性以适应目标的结合位点,因此可能会得到具有更佳抗疟活性的杂合分子。Sunny等[26]主要分析了22种化合物的构效关系,化合物14为抗疟活性较好的一类化合物(图6)。在14(a)结构中,n=1,R为N-甲基哌嗪;14(b)结构中,n=2,R为N-乙基哌嗪。这两者对氯喹敏感疟原虫以及氯喹耐受疟原虫均具有很高的活性,且选择性都较高。此外,他们还研究了喹啉与嘧啶之间碳链长度对活性的影响,发现碳链长度对活性影响不大。而R基对活性有较大影响,结果显示,活性由强变弱依次为:乙基哌嗪、甲基哌嗪、吗啉和哌啶。在细胞毒性方面,这类化合物大多具有细胞毒性,但是其产生细胞毒性的浓度远高于其发挥抗疟作用的浓度,因此可能可以被用来进行抗疟疾治疗。

图6 喹啉嘧啶类化合物结构Fig.6 The structure of quinoline pyrimidine compounds
4 噻吩并嘧啶类化合物
Gonzalez等[27]经过高通量筛选得到43种噻吩并嘧啶类化合物(图7)。化合物15为其基本结构。经构效关系的研究得出:1)R2上的苯基是必要的;2)R上甲胺基变成异丙胺基之后,活性降低。化合物16为该项研究的起始物,具有一定的抗疟活性(IC50=73 nmol/L),且选择性、溶解性都较好。对化合物16进行结构改造,得到一系列具有高抗疟活性(IC50<20 nmol/L)的化合物(如化合物17~21)。在苯环上引入氰基得到化合物17和18。研究发现:这2个化合物具有更好的活性(IC50分别为17和19 nmol/L),且选择性、溶解性没有太大影响。此外,化合物16、19和20可以抑制拜耳氏小鼠模型的疟原虫活性,并且当以4组50 mg/kg剂量口服给药时,小鼠平均生存时间显著提高。而喹啉并嘧啶类化合物在相应的剂量下未观察到抗疟效应[26]。总体来看:药物动力学研究显示化合物16具有高分布量、静脉给药后中度的血液清除度和适度的生物利用度。
5 总结
含嘧啶骨架的杂环类化合物为抗疟药的研发提供了新思路,其反应活性较好,易于合成及改造,且具有多种药理活性。在抗疟机制方面,嘧啶类化合物以二氢叶酸还原酶或组蛋白甲基转移酶为靶点,发挥着多种机制的抗疟活性。基于此,可与其他抗疟机制的化合物(如,喹啉等)进行合并、改造,实现抗疟活性的最大化,并克服耐药性的产生。此外,高通量筛选技术对药物化学中先导化合物的发现也至关重要,新型抗疟药物的发现还有赖于该技术的不断发展和完善。
参考文献:
[1] RAFAEL L,ALAN D L,MOHSEN N,et al.Global malaria mortality between 1980 and 2010: a systematic analysis[J].Lancet,2012,379:413-431.
[2] RIDIEY R J,Medical need,scientific opportunity and the drive for antimalarial drug[J].Nature,2002,415:686-693.
[3] National Institute of Allergy and Infectious Diseases.Life cycle of the malaria parasite[EB/OL].[2017-11-26.]https://www.niaid.nih.gov/lab-sections/3135.
[4] PAUL R G,CRAIG A H,BRAD E S,et al.Optimization of 2-anilino 4-amino substituted quinazolines into potent antimalarial agents with oral in vivo activity[J].J Med Chem,2017,60(3):1171-1188.
[5] NOEDL H,SE Y,SCHAECHER K,et al.Evidence of artemisinin resistant malaria in western Cambodia[J].N Engl J Med,2008,359(24):2619-2620.
[6] ARIEY F,WITKOWSKI B,AMARATUNGA C,et al.A molecular marker of artemisinin resistantPlasmodiumfalciparummalaria[J].Nature,2014,505(7481):50-55.
[7] ASHLEY E A,DHORDA M,FAIRHURST R M,et al.Spread of artemisinin resistance inPlasmodiumfalciparummalaria[J].N Engl J Med,2014,371(5):411-423.
[8] PLOUFFE D,BRINKER A,MCNAMARA C,et al.In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-through put screen[J].Proc Natl Acad Sci USA,2008,105(26):9059-9064.
[9] MALMQUIST N A,MOSS T A,MECHERI S,et al.Small-molecule histone methyltransferase inhibitors display rapid antimalarial activity against all blood stage forms inPlasmodiumfalciparum[J].Proc Natl Acad Sci USA,2012,109(41):16708-16713.
[10] TRELLE M B,SALCEDOAMAYA A M,COHEN A M,et al.Global histone analysis by mass spectrometry reveals a high content of acetylated lysineresidues in the malaria parasitePlasmodiumfalciparum[J].J Proteome Res,2009,8(7):3439-3450.
[11] SHI Y,DESPONTS C,DING S,et al.A combined chemical and genetic approach for the generation of induced pluripotent stem cells[J].Cell Stem Cell,2008,2(6):525-528.
[12] SHI Y,DESPONTS C,DO J T,et al.Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds[J].Cell Stem Cell,2008,3(5):568-574.
[13] VEDADI M,BARSYTELOVEJOY D,LIU F,et al.A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells[J].Nat Chem Biol,2011,7(8):566-574.
[14] LIU F,CHEN X,ALLALIHASSANI A,et al.Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent andselective inhibitor of histone lysine methyltransferase G9a[J].J Med Chem,2009,52(24):7950-7953.
[15] LIU F,CHEN X,ALLALIHASSANI A,et al.Protein lysine methyltransferase G9a inhibitors:design,synthesis,and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines[J].J Med Chem,2010,53(15):5844-5857.
[16] LIU F,BARSYTELOVEJOY D,ALLALIHASSANI A,et al.Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines[J].J Med Chem,2011,54(17):6139-6150.
[17] CHANG Y,ZHANG X,HORTON J R,et al.Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294[J].Nat Struct Mol Biol,2009,16(3):312-317.
[18] CHANG Y,GANESH T,HORTON J R,et al.Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases[J].J Mol Biol,2010,400(1):1-7.
[19] YUTHAVONG Y,TARNCHOMPOO B,VILAIVAN T,et al.Malarial dihydrofolatereductase as a paradigm for drug development against a resistance-compromised target[J].Proc Natl Acad Sci USA,2012,109(42):16823-16828.
[20] SRIVASTAVA A,DUDLEYA A,LUKENS A K,et al.Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate[J].Nat Commun,2015,6:6715.
[21] HARGREAVES S L,PILKINGTON B L,RUSSELL S E,et al.The synthesis of substituted pyridylpyrimidine fungicides using palladium catalysed cross-coupling reactions[J].Tetrahedron Lett,2000,41(10):1653-1656.
[22] LI Y X,LUO Y P,XI Z,et al.Design and syntheses of novel phthalazin-1(2H)-one derivatives asacetohydroxyacid synthase inhibitors[J].J Agric Food Chem,2006,54(24):9135-9139.
[23] REGNIER G L,CANEVARI R J,LE DOUAREC J C,et al.Triphenylpropyl-piperazine derivatives as new potent analgetic su-bstances[J].J Med Chem,1972,15(3):295-301.
[24] SONDHI S M,SINGH N,JOHAR M,et al.Synthesis,anti-inflammatory and analgesic activities evaluation of some mono,bi-and tricyclic pyrimidine derivatives[J].Bioorg Med Chem,2005,13(22):6158-6166.
[25] GALMARINI C M,MACKEY J R,DUMONTET C,et al.Nucleoside analoguesand nucleobases in cancer treatment[J].Lancet Oncol,2002,3(7):415-424.
[26] SUNNY M,RAJESH U C,KHAN S I,et al.Novel 4-aminoquinoline-pyrimidine based hybrids with improved in vitro and in vivo antimalarial activity[J].ACS Med Chem Lett,2012,3(7):555-559.
[27] GONZALEZ C D,LE M C,DOUELLE F,et al.2,4-Diaminothienopyrimidines as orally active antimalarial agents[J].J Med Chem,2014,57(3):1014-1022.
