高三酰甘油血症对急性坏死性胰腺炎大鼠腺泡细胞自噬的影响
2018-05-07梅启享曾悦陆颖影
梅启享 曾悦 陆颖影
Fundprogram:National Natural Science Foundation of China(81200322,81600500)
急性胰腺炎(AP)是临床常见且预后较差的疾病之一,它的发病机制一直属于研究热点。目前多认为它的发生是由于胰酶酶原在胰腺腺泡细胞内的提前激活所致,而胰腺腺泡细胞内胰蛋白酶原激活被认为与自噬紊乱有关。作为AP中胰腺腺泡细胞的早期事件,自噬活性的调节可影响AP病情的严重程度[1]。高三酰甘油血症在胰腺炎发病因素中名列第3位,是促进胰腺炎重症化的一个重要因素[2]。有研究发现自噬可能参与高脂诱导的疾病进程[3]。然而目前高脂因素对于胰腺腺泡细胞自噬影响的研究尚属空白。自噬相关基因(autophagy-related gene,ATG)蛋白的表达在自噬的发生发展过程中尤为重要,其中定位于自噬体膜的微管相关蛋白轻链3(microtubule associated protein light chain 3, LC3)、与自噬溶酶体功能相关的溶酶体相关膜蛋白2(lysosome associated membrane protein 2,LAMP-2)以及识别降解底物相关的p62被认为是自噬作用的标志物,它们的改变预示了自噬活性的改变[4]。本研究观察AP及高脂血症相关性AP胰腺腺泡细胞自噬的变化,探讨高脂对AP时自噬活性的影响。
材料和方法
一、动物模型制备和实验分组
40只雄性SD大鼠,SPF级,体重200~220 g,购自上海斯莱克实验动物有限公司。按数字表法随机分为假手术对照组、急性坏死性胰腺炎(acute necrotizing pancreatitis, ANP)组、高脂血症(hyperlipidemia,HL)组、高脂血症性ANP(hyperlipidemia ANP,HANP)组,每组10只。对照组给予正常饮食喂养;ANP组给予正常饮食喂养2周后采用胰胆管逆行注射3.5%牛黄胆酸钠溶液的方法制备ANP模型;HL组给予高脂饲料(正常饲料77%+猪油20%+胆固醇3%)喂养2周;HANP组给予高脂饲料喂养2周后同上述方法制备ANP模型。对照组和HL组腹腔内注射等容积生理盐水。各组大鼠于喂养2周后尾静脉采血,分离血清,置-20℃保存。造模后48 h处死大鼠,腹主动脉采血,分离血浆,置-20℃保存;取胰腺组织,部分立即置液氮保存,部分用10%甲醛固定备用。
二、方法
1.血三酰甘油(TG)、总胆固醇(TC)、淀粉酶水平检测:血标本送上海市第一人民医院生物化学实验室检测TG、TC及淀粉酶水平。
2.胰腺组织病理检查:取固定的胰腺组织,常规脱水、石蜡包埋、切片及HE染色,光镜下读片,并参照Schmidt等[5]的标准对胰腺组织进行病理评分。
3.胰腺组织LC3Ⅰ、LC3Ⅱ、p62、LAMP-2蛋白检测:应用RIPA裂解液提取胰腺组织总蛋白,应用BCA试剂盒(碧云天生物技术有限公司)测定蛋白浓度。采用蛋白质印迹法检测LC3Ⅰ、LC3Ⅱ、p62、LAMP-2蛋白表达量,以GAPDH为内参。兔抗大鼠LC3Ⅰ、LC3Ⅱ购自CST公司,兔抗大鼠p62购自Proteintech公司,兔抗大鼠LAMP-2购自Abcam公司,工作浓度分别为1∶1 000、1∶1 000、1∶2 000;羊抗兔IgG-HRP二抗购自碧云天公司,工作浓度1∶2 000。最后ECL(Millipore公司)化学发光,X片曝光成像。应用天能Tanon 5200全自动化学发光成像分析系统扫描,以目的条带与内参条带的灰度值比表示蛋白相对表达量,并计算LC3Ⅱ/Ⅰ比值。
4.胰腺腺泡细胞自噬体及自噬溶酶体观察:取液氮保存的胰腺组织标本,切取大约1 mm×1 mm×1 mm立方体,洗去残留的血液和组织液后迅速放入新鲜3%的戊二醛(pH7.4)固定4 h以上,而后置入 1%的锇酸固定液。常规按电镜切片方法行超薄切片,厚度约70 nm。经醋酸双氧铀和柠檬酸铅双染色,在日本JEM1230透射电镜下随机选取10个不同视野,观察并记录每个视野内自噬体和自噬溶酶体的数量,取均值。
三、统计学处理

结果
一、血清TG、TC、淀粉酶水平的变化
对照组、HL组、ANP组、HANP组血清TG水平分别为(0.36±0.17)、 (0.72±0.37)、(0.28±0.13)、(0.82±0.49)mmol/L,TC分别为(1.54±0.25)、(2.07±0.45)、(1.70±0.18)、(1.97±0.06)mmol/L。两高脂饲料喂养组大鼠的TG、TC水平均显著高于两正常饲料喂养组,差异均有统计学意义(TG:t值分别为3.455、3.288,P值均<0.01;TC:t值分别为2.916、4.502,P值均<0.05),表明高脂血症大鼠模型成功建立。
对照组、HL组、ANP组、HANP组血淀粉酶活性分别为(1 818±167)、(1 895±132)、(4505.60±549.16)、(4 830±328)U/L,对照组与HL组的差异无统计学意义,ANP和HANP组则较对照组显著升高,差异有统计学意义(t值分别为12.417、14.012,P值均<0.01),提示ANP造模成功。
二、胰腺组织病理学改变
对照组和HL组的胰腺组织无明显病理改变,病理评分基本为0分。ANP组和HANP组胰腺组织均出现不同程度的水肿、出血、坏死和炎性浸润(图1)。ANP组胰腺组织病理评分显著高于对照组,HANP组胰腺病理评分显著高于HL组及ANP组,差异均有统计学意义(P值均<0.01,表1)。
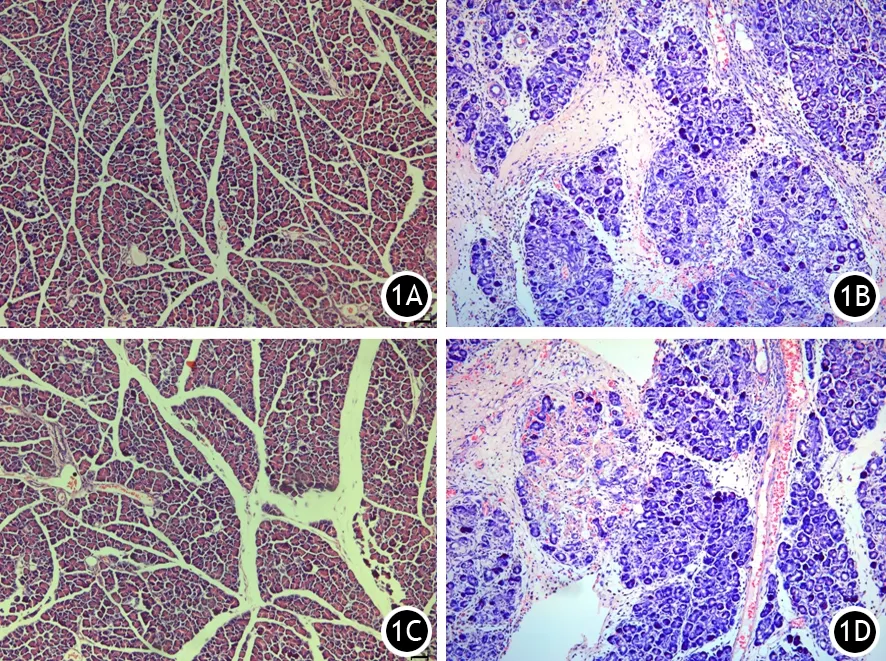
图1 对照组(1A)、ANP组(1B)、HL组(1C)、HANP组(1D)大鼠胰腺组织病理改变(HE ×200)

组别只数总评分水肿出血坏死炎症浸润对照组100.20±00.20±000.45 0.45 HL组100.40±0.20±0.20±000.540.45 0.45ANP组105.60±2.20±0.90±1.10±1.40±0.54a0.28 0.420.420.42HANP组108.00±2.90±1.60±1.40±2.20±0.71bd0.42c0.42c0.42c0.76c
注:与对照组比较,aP<0.01;与HL组比较,bP<0.01;与ANP组比较,cP<0.05,dP<0.01
三、胰腺组织的自噬相关结构改变
根据细胞内自噬体的数量可以判断自噬的活性。透射电镜观察显示,对照组细胞核呈卵圆形,细胞核膜结构完整,胞质中细胞器形态正常,未见自噬体,自噬活性较弱。ANP组细胞核呈不规则形,线粒体有轻度肿胀,可见“C”形的双层膜结构样的自噬小体及包含降解物的自噬体,并出现自噬小体和溶酶体融合现象,溶酶体内可见吞噬内容物,自噬活性大大增强。HL组细胞核呈卵圆形和不规则形,细胞连接紧密,细胞器结构较完整,有疑似自噬小体,自噬活性较弱。HANP组细胞核呈不规则形,染色质部分凝集,细胞器形态较完整,可见分泌颗粒,有疑似自噬小体,自噬活性相对ANP组受到抑制(图2)。
对照组、ANP组、HL组、HANP组的自噬体数量分别为(0.30±0.58)、(4.00±1.00)、(0.67±0.58)、(1.70±0.58)个,ANP组显著多于其他3组,差异均有统计学意义(P值均<0.05)。
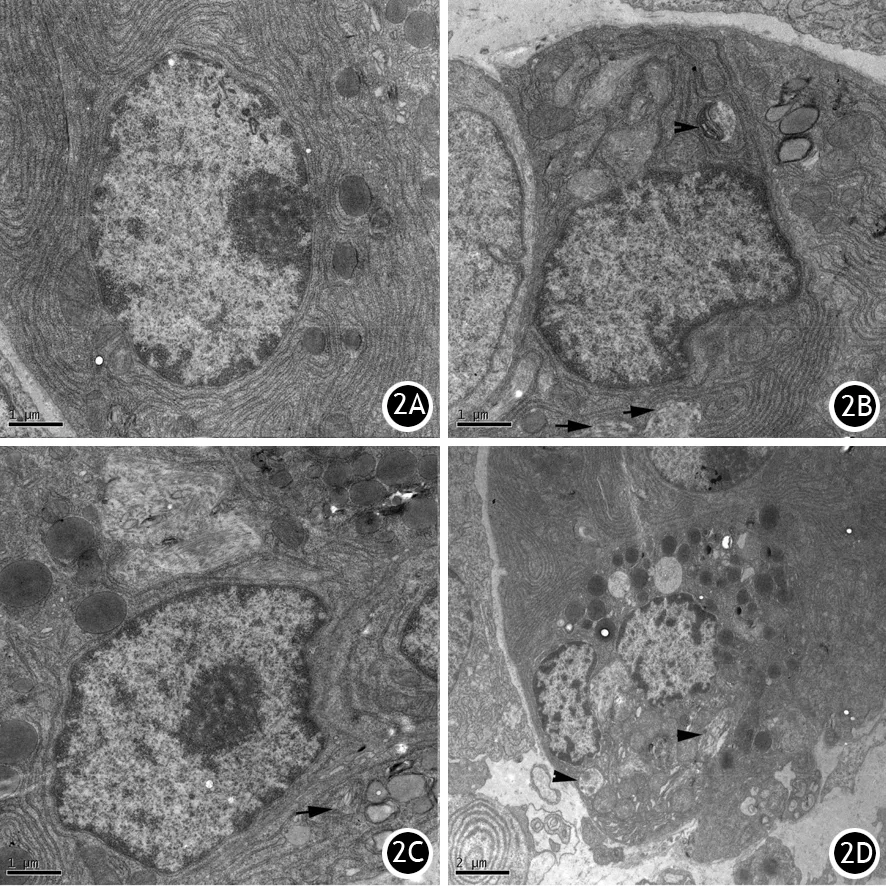
图2 对照组(2A)、ANP组(2B)、HL组(2C)、HANP组(2D)大鼠胰腺腺泡细胞内自噬体及自噬溶酶体(A、B、C×10 000,D×5 000)
四、胰腺组织自噬相关蛋白表达的变化
对照组大鼠胰腺组织p62、LC3Ⅰ、LC3Ⅱ低表达,LAMP-2高表达。ANP组大鼠胰腺组织p62表达及LC3 Ⅱ/Ⅰ比值较对照组增高,LAMP-2表达降低,差异均有统计学意义(t值分别为9.062、7.737、3.619,P值均<0.05)。HSAP组大鼠胰腺组织p62表达及LC3Ⅱ/Ⅰ比值较HL组和ANP组均降低,差异均有统计学意义(t值分别为23.35、6.801、4.187、19.44,P值均<0.01),LAMP-2表达与HL组和ANP组差异均无统计学意义(表2,图3)。

表2 各组LC3Ⅱ/Ⅰ比值及p62、LAMP-2蛋白表达
注:与对照组比较,aP<0.05;与HL组比较,bP<0.01;与ANP组比较,cP<0.01
讨 论
高脂血症作为AP病因仅次于胆源性和酒精性因素,高脂血可使胰腺炎趋向重症化,然而其具体机制尚未完全阐明[6]。Guo等[7]报道,在3T3-L1脂肪细胞的分化过程中可以通过靶向调节自噬来影响细胞的分化,表明自噬可能与肥胖、脂肪代谢紊乱有关。Dall′Armi 等[8]认为脂质和脂质代谢酶可以通过控制哺乳动物TOR(mTOR)途径上的信号级联来负调节自噬的启动。

图3 对照组(1)、ANP组(2)、HL组(3)、HANP组(4)胰腺组织蛋白表达
自噬通过对细胞器的有效调控可以保持胰腺腺泡细胞的内环境稳定和正常分泌功能[9]。Diakopoulos等[10]通过构建自噬相关基因ATG-5缺陷的小鼠证明自噬对于胰腺的稳态至关重要,认为自噬的损伤是胰腺炎致病的关键。异常自噬的发生可使得胰蛋白酶原在自噬溶酶体内被加快激活为胰蛋白酶,使胰腺腺泡空洞聚集,并且导致胰酶酶原的提早活化,从而加重胰腺炎。自噬是脂质代谢的重要途径之一,自噬活性的降低使脂质代谢功能障碍,促使脂质蓄积。然而,尚无有关高脂血症与AP中自噬相关性机制研究的报道。
溶酶体膜内的LAMPs是一种高度糖基化的跨膜蛋白, 在保持溶酶体结构和功能方面扮演着重要角色。它可使细胞免受酸性水解,并参与调控溶酶体与自噬体的融合,对自噬进程有重要影响。溶酶体相关膜蛋白2(LAMP-2)被认为是自噬体与溶酶体的降解和融合的重要蛋白。Zhu等[11]通过注射精氨酸制备AP模型,发现LAMP-2的表达下降,认为溶酶体与自噬体的融合障碍是AP异常自噬发生的原因。LC3是自噬体形成过程中的重要功能蛋白,在自噬启动中发挥着重要作用。它存在着两种互相转化的形式,即LC3-Ⅰ和LC3-Ⅱ。LC3 Ⅱ定位于自噬囊泡体膜上,LC3Ⅱ/Ⅰ的比值可以反映自噬的活性。在自噬降解过程中,p62/SQSTM1是自噬降解能力的重要评价指标[12]。因此,LC3、p62、LAMP2的表达变化可以代表自噬活性的改变。
本实验研究结果显示,ANP组自噬相关蛋白LC3、p62表达增加,LAMP-2表达下降,证明胰腺炎症反应中自噬增强,且自噬溶酶体功能发生了障碍,HANP组LC3Ⅱ/Ⅰ比值、p62表达较ANP组下降,提示胰腺自噬活性降低,表明在高脂因素影响下,自噬活性被抑制。但HANP组与ANP组LAMP-2表达未见明显差异,提示高脂因素的作用可能与溶酶体对自噬的调控作用无显著相关性。
研究发现,AP时胰腺腺泡细胞出现自噬小体,并随着胰腺炎的病情程度加重,自噬的结构发生改变,自噬小体体积增大,数量增多,出现自噬小体和溶酶体的融合现象[13]。本研究通过透射电镜观察,ANP组可见“C”形的双层膜结构样的自噬小体及包含降解物的自噬体,出现自噬小体和溶酶体融合现象,HANP组有疑似自噬小体,自噬活性发生了改变。HANP组的胰腺病理损伤较对照组和ANP组更严重,然而,它的自噬活性被相对抑制,提示高三酰甘油血症可能参与了AP异常自噬的发生发展过程,并进一步导致疾病重症化。
[1] Gukovsky I, Gukovskaya AS. Impaired autophagy underlies key pathological responses of acute pancreatitis [J]. Autophagy, 2010, 6(3): 428-429.DOI: 10.4161/auto.6.3.11530.
[2] Christian JB, Bourgeois NE, Lowe KA. Cholesterol screening in US adults and awareness of high cholesterol among individuals with severe hypertriglyceridemia: National Health and Nutrition Examination Surveys 2001-2008 [J]. J Cardiovasc Nurs, 2015, 30(1): 26-34. DOI: 10.1097/JCN.0000000000000101.
[3] Liang L, Shou XL, Zhao HK, et al. Antioxidant catalase rescues against high fat diet-induced cardiac dysfunction via an IKKbeta-AMPK-dependent regulation of autophagy [J]. Biochim Biophys Acta, 2015, 1852(2): 343-352. DOI: 10.1016/j.bbadis.2014.06.027.
[4] Sun A, Wei J, Childress C, et al. The E3 ubiquitin ligase NEDD4 is an LC3-interactive protein and regulates autophagy [J]. Autophagy, 2017, 13(3): 522-537.DOI: 10.1080/15548627.2016.1268301.
[5] Schmidt J, Rattner DW, Lewandrowski K, et al. A better model of acute pancreatitis for evaluating therapy [J]. Ann Surg, 1992, 215(1): 44-56.
[6] Zheng J, Wu J, Chen J, et al. Therapeutic effects of quercetin on early inflammation in hypertriglyceridemia-related acute pancreatitis and its mechanism [J]. Pancreatology, 2016, 16(2): 200-210. DOI: 10.1016/j.pan.2016.01.005. Epub 2016 Jan 28.
[7] Guo L, Huang JX, Liu Y, et al. Transactivation of Atg4b by C/EBPbeta promotes autophagy to facilitate adipogenesis [J]. Mol Cell Biol, 2013, 33(16): 3180-3190.DOI: 10.1128/MCB.00193-13.
[8] Dall′armi C, Devereaux KA, Di Paolo G. The role of lipids in the control of autophagy [J]. Curr Biol, 2013, 23(1): R33-R45. DOI: 10.1016/j.cub.2012.10.041.
[9] Gukovskaya AS, Pandol SJ, Gukovsky I. New insights into the pathways initiating and driving pancreatitis [J]. Curr Opinion Gastroenterol,2016.DOI:10.1097/MOG.0000000000000301.
[10] Diakopoulos KN, Lesina M, Wormann S, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes [J]. Gastroenterology, 2015, 148(3): 626-638.e17.DOI: 10.1053/j.gastro.2014.12.003.
[11] Zhu H, Yu X, Zhu S, et al. The fusion of autophagosome with lysosome is impaired in L-arginine-induced acute pancreatitis [J]. Int J Clin Exp Pathol, 2015, 8(9): 11164-11170.
[12] Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis [J]. Mol Cells, 2013, 36(1): 7-16. DOI: 10.1007/s10059-013-0140-8. Epub 2013 May 24.
[13] Yu C, Yu X, Zhu HW, et al. Expression pattern of HMGB1 and its association with autophagy in acute necrotizing pancreatitis [J]. Mol Med Rep, 2016, 14(6): 5507-5513. DOI: 10.3892/mmr.2016.5945.
