微吸管单类细胞收集法对哺乳动物内耳细胞转录组的研究
2018-04-19李轶刘会占何志洲
李轶刘会占何志洲
1首都医科大学附属北京同仁医院院耳鼻咽喉头颈外科,耳鼻咽喉头颈科学教育部重点实验室首都医科大学(北京100730)2Department of Biomedical Sciences,Creighton University SchoolofMedicine
对于多细胞生物体来说,虽然每个细胞的基因组是相同的,但是每个细胞表达的基因(即转录组)各不相同。转录组是指一个细胞中的所有转录本,包括mRNAs、rRNAs、tRNAs以及其它非编码RNA,它控制着不同细胞所表现出的功能和形态。转录组在单细胞水平被调节,甚至在同一类细胞中,转录组也会随时间、环境有所不同。研究不同发育阶段、生理、病理条件下的转录组对揭示细胞分化、增殖、衰老、代谢、病理变化、形态和功能特点具有重要意义。
哺乳动物的听觉感受器(Corti’s器)由内、外毛细胞和支持细胞构成,内毛细胞是感受器细胞,外毛细胞是效应器细胞[1]。支持细胞包括内指细胞、柱细胞、Deiters'细胞、内缘细胞、Hensen细胞和Claudius细胞,为毛细胞提供必需的代谢、电解质环境以维护其正常生理功能,并为Corti’s器的机械特性提供支撑[2]。毛细胞和支持细胞共同来源于听囊内的感觉前体细胞。在非哺乳动物如鱼类、两栖类和鸟类,支持细胞在毛细胞凋亡后可自发的转分化为毛细胞[3]。新生哺乳动物的内指细胞、柱细胞在毛细胞凋亡后也能转分化为毛细胞[4,5]。成年哺乳动物的支持细胞则丧失这种能力[6],因此分析内耳Corti’s器各细胞的转录组对研究哺乳动物毛细胞再生的分子机理和寻找基因疗法的靶点具有重要指导意义。
在单类细胞转录组的研究中,最大的困难是获取足够数量的高纯度单一细胞。现有的获取单类细胞样本的方法很难在内耳组织中加以应用。这是由于哺乳动物耳蜗组织的特殊性:细胞本身数量较少、分离困难、难以进行体外培养,另外,成年动物的毛细胞易损伤,在体外只能存活很短的时间。因此对于成年哺乳动物耳蜗内的各类细胞,很难获得数量足够的高纯度的一类细胞来进行转录组研究[7]。目前听觉领域的此类研究大都局限在新生和胚胎动物的毛细胞和支持细胞[8-10]。
采用微吸管单类细胞收集技术,可以实现对内耳细胞进行单个分类收集,并获得足够的样本量。微吸管细胞收集技术是将动物的耳蜗解剖出来后进行消化,得到Corti’s器中各种单个离体活细胞,运用微操纵器连接玻璃微电极在显微镜直视下,根据各种细胞的形态特征,用负压吸引、收集单种活细胞的技术[11-13]。采用这种方式收集内耳细胞,使分析成年动物的内耳单类细胞转录组成为可能[13,14]。
本文以成年小鼠为例,详细介绍了耳蜗毛细胞和支持细胞的分离方法、辨认特征,以及采用微吸管收集单类细胞的设备和方法,该方法同样适用于低等动物如鸟类和鱼类(斑马鱼)内耳毛细胞的分离和收集。同时详细介绍了进行转录组研究所采用的单类细胞RNA收集、提取、放大和逆转录的方法。
1 材料和方法
1.细胞的获取和分离:使用CBA/J小鼠(25至30天龄,购买于美国Jackson Laboratory),性别不限。将基底膜与Corti’s器一起分离并转移到含有酶消化培养液的小培养皿中(图1-A,1)。酶消化培养液含有1ml Leibovitz’s-15(L-15)和1mgIV型胶原酶(Sigma)。在室温下孵育5分钟后,将组织用小吸管转移到含有无酶培养液(L-15,7.35 pH,300mOsm)的小液槽(体积为0.8-1.0ml)中(图1-A,2)。用小号移液管轻轻吹打组织以使细胞分离。然后将装有毛细胞的小液槽(图1-A,2)安放在装有摄像机的倒置显微镜(Olympus IX71)的镜台上(图1-B)。
2.分离细胞的收集:为了收集孤立的细胞,使用直径为30μm左右的两个辅助微吸管来接收和转移细胞。微吸管由微电极拉制仪(Model P-97,Sutter Instrument)用直径为1.5mm薄壁玻璃管拉制而成,尖端加热抛光(Micro Forge MF-830,Nar⁃ishige)防止损伤细胞。微吸管分别安装在两台Nar⁃ishige显微操作器(MHW-3,Narishige,图1-B,白色箭头所指)上的两个独立的电极支架中(图1-B,黄色箭头所指)。通过移动显微操作器和显微镜载物台,可将要收集的细胞定位在微吸管尖端附近。微吸管的另一端通过电极支架的出口与2mm直径的硅胶管相连,并连接到微米螺旋推动式注射器(图1-C),以提供正压或负压,以便吸取或排出细胞。图2显示的是一个外毛细胞在被吸入微吸管之前(图2-A)和之后(图2-B)的显微镜下照片。微吸管内充满细胞外液,当4至5个细胞吸入玻璃微吸管后,将微吸管从液槽中取出并迅速转移到含有50μlRNAlater(Thermo Fisher Scientific,Waltham,MA)的微量离心管的盖子里(图1-A,3),并通过施加正压将细胞从移液管中排出。微量离心管要放置在冰块中以防止细胞收集期间RNA的降解。约500个以上细胞所获得的总RNA可满足高质量RNA测序分析的要求,重复这些步骤,直到采集到足够的细胞数。在正常情况下,我们可以从每只小鼠中收集到约50个内毛细胞和100-200个外毛细胞,时间在一小时内。
为了获得高纯度的细胞,常采取以下方式以避免所采集的样本被其他类型的细胞污染。首先,要确定收集的细胞种类。哺乳动物的内毛细胞、外毛细胞和支持细胞都具有独特的形态特征,很容易根据其形态识别[11]。对于任何形态特征不明确的细胞不予以收集。其次,要确保所收集的细胞表面没有附着任何其他类型的细胞。第三,注意施加在微吸管上的吸力,以避免吸入杂细胞,在微吸管移出培养液的瞬间要保持压力恒定防止吸入过多的液体和其他细胞。
3.RNA提取和纯化和反转录:将所获得的样本拿到专门处理RNA的房间,在专门处理RNA的超净台上进行RNA提取。使用Qiagenmicro RNA isolation kit进行细胞裂解、总RNA提取和收集,所收集的RNA不要存放,使用Qubit@RNA HSAssay试剂盒在Qubit®2.0荧光计上进行RNA数量的检测,使用Agilent RNA 6000 Pico试剂盒,在Agilent 2100生物检测仪器上测定RNA的质量,若样本存在少量DNA污染,可采用DNA酶消化去除污染。由于从内耳细胞中可获得的RNA总量相对较少,因此获得较高质量的RNA后,建议尽快将RNA进行反转录和扩增,以保证RNA不会因降解造成质量的降低。使用Colontech Laboratories cDNA合成试剂盒(SMARTer@UltraTM Low input RNA for illu⁃mine@sequencing-HV kit)将所获得的RNA反转录成cDNA,并进行cDNA的扩增和纯化。同样,使用Qubit®2.0仪器对扩增及纯化后所获得的cD⁃NA进行样本数量检测,使用Agilent High Sensitity DNA试剂盒来检测cDNA的质量。然后将可所获得的样本进行转录组测序。对每种采集的细胞我们都分别制备三个样本,以保证样本的生物重复性和可靠性。
2 结果
每种单类细胞收集500个左右即可满足转录组分析的需要,视细胞种类不同约需7-10只小鼠,每种细胞均进行三个生物学重复。
2.3.3 5-HT 3RA的应用情况 本研究中患者应用的 5-HT3RA 包括 Tro、Pal和 Ond。“指南”推荐 Tro 只在第1天静脉应用或口服5mg。有59例(53.15%)连续多日应用Tro,疗程过长。应用Pal存在的主要问题与Tro一致,有11例(9.91%)应用Pal疗程过长。有4例将Pal与Tro重复应用,因药物与受体的结合存在饱和现象,重复用药并不能增强疗效,反而会增大不良反应的发生率,两药重复应用为不合理用药。Ond应用主要存在超致吐级别应用、给药剂量偏小和疗程不合理。Ond为短效的5-HT3RA,应用Ond的例数较Pal和Tro少。见表6。
1、内耳细胞的形态学特征及辨认:小鼠的内毛细胞、外毛细胞、柱细胞和Deiters'细胞在分离后常常保持了它们特有的形态学特征,在光学显微镜下容易辨认,下面列举了一些图例来描述它们的形态学特征。图2-C和图2-D中所显示的是分离得到的成年小鼠单个内毛细胞、外毛细胞、柱细胞和Deiters'细胞在光镜下(使用DIC镜头)的照片,图2-E显示的是鸡的长细胞(tall hair cell)和短细胞(shorthair cell)的照片,长细胞的功能与哺乳动物内毛细胞类似,短细胞的功能与外毛细胞功能类似。图2-F显示的是斑马鱼内耳的毛细胞的照片。哺乳动物内毛细胞的形状通常被描述为烧瓶状,在表皮板和胞体之间有一个狭窄的颈部,细胞核位于细胞中下部。小鼠内毛细胞的直径约8-10μm,长度约15-20μm。外毛细胞呈圆柱形,细胞核位于细胞底部,小鼠外毛细胞的直径约6μm,长度约15-32μm,两类毛细胞顶端都有约4-5μm长的纤毛,纤毛是毛细胞特征性标记。另外,两类毛细胞都具有丰富的线粒体(在光镜下呈颗粒状并具有明显的布朗运动)。柱细胞呈柱状,可以明显的看见胞浆内纵行排列的丝状结构即Actin,表皮板端膨大并没有纤毛,Deiters'细胞具有特征性的指突,如图2-D中黑色箭头所示,还具有典型的容纳外毛细胞突触端的杯状结构(图2-D中白箭头所示)。所获得的单细胞还可根据不同实验目的用来进行分子生物学、单细胞电生理、形态学实验。图3-A所显示的是使用扫描电镜研究单个外毛细胞的形态的扫描电镜照片,图3-B显示的是使用电压钳技术记录外毛细胞离子通道的光镜照片。
2、使用微吸管单类细胞收集法所获取的单类细胞总RNA样本的可靠性:从采集到的单类细胞样本中提取出的总RNA,我们进行了的RNA样品的浓度以及质量的检测。根据我们的经验,RNA样本的浓度至少要保证在100ng/ml以上才能满足后续实验要求,我们以此法所获得的单类细胞总RNA样本浓度一般都可以达到4μg/ml左右。为了保证能在RNA-seq检测中可以测到尽可能完整的mRNA序列,还需对总RNA样本进行质量检测。RNA的质量判断包括RNA完整性及纯度两个方面,在检测RNA样本所获得的电泳图中,如果28S和18S条带清晰,且亮度比28S:18S>1,则证明该样本的完整性很好,基本无降解。同时,点样槽内或附近不应存在其他荧光区带,否则表明RNA样品中有DNA污染。图4-A中显示的是我们以此方法获取的小鼠外毛细胞总RNA样本(使用Agilent RNA 6000 Pico试剂盒,在Agilent2100生物分析仪上测定)的质量,可以看到18S和28S这两个核糖体RNA的电泳峰分别出现在38秒和46秒,同时两峰呈现高、尖的形态,说明RNA降解少、完整性好。Agilent2100生物检测仪器自带分析软件,根据信号面积、强度、比值等信息计算RNA完整值(RIN)对RNA完整度进行评估,图中所显示的样本检测后的RIN值为9.2(10为满分)。通常选择用于测序的RNA样品的RIN值至少要大于8.0,我们用于测序的总RNA样本的RIN都为9.0以上。
同样,在进行RNA-seq的过程中,也需要一些指标来验证测序样本的质量,以保证测序结果的可靠性。首先,数据分析的第一步,要把测到mRNA片段,比对(Mapping)到基因组上,验证测序中所捕获到的mRNA是否完整。图4-B上方显示的是成年小鼠外毛细胞RNA测序样本中捕获的mRNA片段在参考基因上的分布均匀性统计图,可见图形的5’端的曲线非常饱满,高度和3’端几乎相等,中间区域分布均匀,无偏向性分布,这表明测序中所捕获的mRNA大部分是完整的。图4-B中下方显示的是对于所捕获的mRNA样本统计数进行基因组比对后的分布情况(Alignment Distribution)。图4-C中显示的是所捕获的mRNA样本统计数进行基因组比对后总体情况,能够比对上的样本统计数的比例是一个重要的质量控制参数,这个参数能指示整个测序的准确性和是否有污染DNA的存在。一般情况下,样本统计数的比对率能达到90%以上就表明测序样本具有相当高的质量,当使用转录组作为参考时,一般还会将这个参数下调。从图中可见我们所获得标本的总比对率高达95.24%。
3、转录组数据获取及利用单类细胞转录组数据信息进行数据分析:RNA-Seq数据分析时使用TopHat v1.4.1将样品分别定位到小鼠基因组(mm10,GRCm38)。使用 HTSeq-count从配对序列中得到基因表达,使用DESeq来识别差异表达的基因,假发现率(FDR)设定在5%,至少有2倍的变化阈值,归一化读取计数设置为10%的中位数标准化阅读计数。Entrez Gene,HGNC,OMIM和Ensembl数据库用于验证、参考和分析。
我们已经使用微吸管收集法和RNA-seq技术成功获得了成年小鼠内、外毛细胞、柱细胞、Deit⁃ers'细胞的转录组数据(所有数据均已上传,可在SHARED Harvard Inner-Ear Laboratory Database:https://shield.hms.harvard.edu/index.htm l查阅)。通过对内耳细胞转录组数据的分析,我们可以获得很多重要信息。除了分析哪些基因在这几类细胞中共同表达、差异表达、特异性表达之外,我们也对比分析了耳聋基因在内、外毛细胞、柱细胞和Deiters'细胞上表达情况。
目前已知大约有120个基因的突变或缺陷与遗传性综合征性耳聋或非综合征性耳聋的发生有关[15],去除X染色体连锁和线粒体遗传基因后,我们对其中101个基因的表达情况进行了分析比较。以确定他们在内、外毛细胞、柱细胞和Deiters细胞中表达情况的差异。图5显示了这些耳聋相关基因在内毛细胞、外毛细胞、柱细胞、Deiters细胞中的表达水平。在所有四种细胞类型中,有7个基因(Hgf,Gjb3,Mir96,Col4a6,Foxi1,Myo1a和Pax3)没有检测到表达,有5个基因(Tecta,Esrrb,Diap3,Col4a3和Edn3)的表达水平非常低,或者只在其中一到两种细胞类型中可以检测到表达。在毛细胞中表达的大多数基因也在柱细胞和Deiters细胞中表达,但它们中的大部分表达水平不同。这些基因在毛细胞和支持细胞中的表达谱为了解人类听力损失的病理机制提供了重要的分子基础。

图1 使用微吸管收集细胞所用的设备、装置。Fig.1 Setup for using suction pipette technique for collecting isolated hair cells and supporting cells.
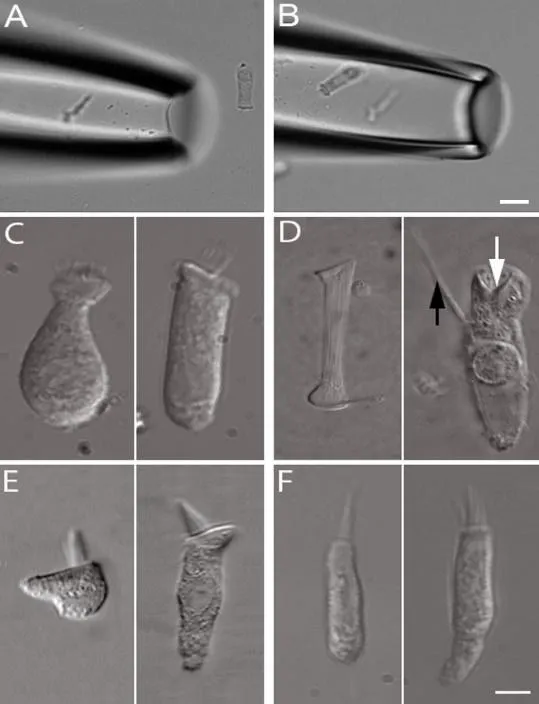
图2 使用微电极收集毛细胞及光镜下内耳各类细胞的图例。Fig.2 Imagesof using suction pipetteand isolated hair cells.
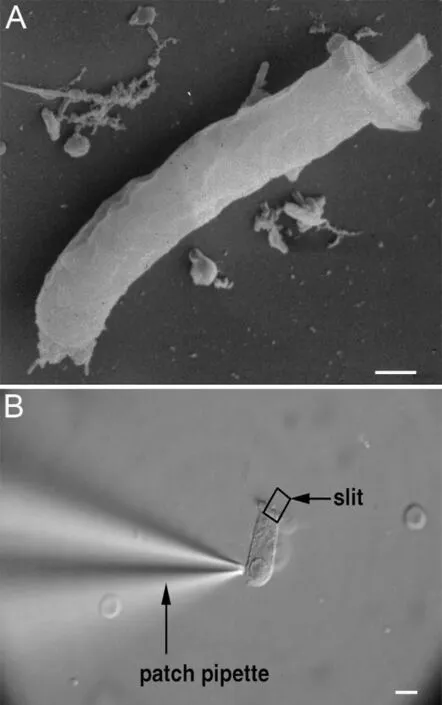
图3 游离的单个外毛细胞Fig.3 Imagesof isolated outerhair cells
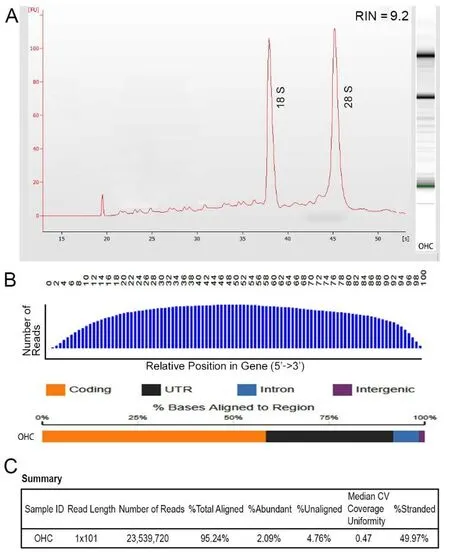
图4 从小鼠外毛细胞提取的RNA质量分析及RNA-seq质量检测结果。Fig.4:Quality analyses of RNA extracted from mouse outer hair cells.
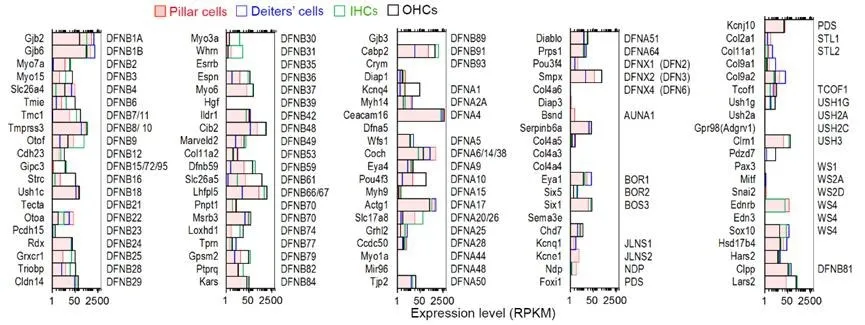
图5 除外X染色体连锁和线粒体遗传基因后,101个耳聋相关基因在内毛细胞、外毛细胞、柱细胞、Deiters细胞中的表达水平.(数据为三个生物学重复的均值)Fig.5 Expression(RPKM)values of 101 deafness-related genes in IHCs,OHCs,pillar cells and Deiters’cells from adultmouse cochleae.(the datawere from means of three biological repeats foreach cellpopulation)
3 讨论
单一种类细胞的异质性对其功能、形态具有至关重要的影响,而大量异质细胞群体的分析只能提供关于群体的平均数据,某些相对微小但潜在相关的重要信息会在群体的数据背景下丢失,因此单类细胞分析在生命科学和生物医学研究的各个领域具有越来越重要的作用[16,17]。为了实现单细胞分析,需要首先进行单细胞分选、收集以获得足够的单类细胞样本。目前,单类细胞的分离面临的主要挑战是分离的产量和质量,或换句话说,所分离细胞的完整性、纯度以及单类细胞分离方法的效率[18,19]。目前被广泛采用的有以下三类单细胞分选方法:1、使用流体技术的分选方法如:流式细胞荧光分选技术(Fluoresence activated cellsorting,FACS);2、使用激光捕获显微切割技术(Laser capturemicrodissection technique LCM);3、人工细胞采集/显微操作法(Manual cellpicking)。
流式细胞荧光分选技术(FACS)的工作原理是将荧光染色或标记的单细胞悬液放入样品管中,并用高压压入充满鞘液的流动室。在鞘液的包裹和推动下,细胞被排成单列,以一定速度从流动室喷口喷出,流动室的喷口上配有一个超高频的压电晶体,充电后振动,使喷出的液流断裂为均匀的液滴,待测细胞就分散在这些液滴之中。细胞性质在进入液滴以前已经被测定,如果其特征与被选定的细胞特征相符,则仪器在这个被选定的细胞刚形成液滴时给整个液柱充以指定的电荷,使其形成液滴时带有特定的电荷,而未被选定细胞的细胞液滴和不含细胞的空白液滴不被充电。带有电荷的液滴向下落入偏转板的高压静电场,按照所带电荷符号发生偏转,落入指定的收集器内,完成分类收集[20-23]。
FACS操作相对简便,分选效率高,细胞分选精确度高(99%以上),速度快[20]。先进的流式细胞仪的分选速度可达每秒25000个细胞以上,因此FACS已被广泛应用于基础和临床研究,但是有几个缺点限制了其应用。首先,细胞在机器中的快速流动过程中,机器和非特异性的荧光分子可能损坏所分选的细胞,使分离失败[24]。其次,在FACS分析之前,细胞需要进行单独的处理以产生带有荧光标记的细胞,这个过程非常耗时[18,19,24,25]。上述两个原因可使分选细胞的RNA质量受到影响或发生变化。
激光捕获显微切割技术(LCM),是一种在显微镜可视条件下,直接从冰冻或石蜡包埋组织切片中获取目标细胞的方法[26]。其工作原理是显微镜直视下选择目标细胞,机械臂控制覆有热塑膜(Ethyl⁃ene Vinylacetate,EVA)的塑料帽,放到组织切片上的目标部位,通过低能红外激光脉冲激活热塑膜,瞬间升温可在使EVA膜局部熔化,熔化的EVA膜渗透到切片上极微小的组织间隙中,并在几毫秒内迅速凝固。组织与膜的粘合力超过了其与载玻片间的粘合力,从而可以选择性地转移目标细胞。激光脉冲通常持续0.5~5.0毫秒,并且可在整个塑料帽表面进行多次重复,从而可以迅速分离大量的目标细胞[27]。
LCM最显著的优点在于其迅速、准确和多用途的特性[28]。根据组织结构特点以及所需的切割精确度,来选择激光束的直径大小,可以迅速获取大量的目标细胞,捕获细胞和剩余组织的形态学特征均保持完好,可以较好地控制捕获细胞的特异性[29,30]。但此项技术要求组织表面不能有盖玻片,没有盖片、封片剂和组织间的折射率匹配,干燥组织的折射性质会导致在高放大倍数下模糊细胞细节,使其视觉分辨率受到很大限制,因而无法应用于那些本身缺乏一定结构特点的复杂组织[24]。同时,要进行有效LCM,操作人员必须具有通过显微镜检查形态特征正确鉴定复杂组织中细胞亚群或单细胞的能力。此外,LCM会导致很多技术伪差,包括在制备组织切片期间对细胞的切割,以及激光切割能量对DNA或RNA进行紫外线损伤[18,31]。同时,激光会产生约1μm的切割宽度。因此,解剖靶细胞同时要避免相邻细胞或其他非特异性片段的污染,否则会导致假阳性和假阴性。解剖太保守会造成不能包括完整的整个细胞,导致用于下游分析的RNA收获不足,但是解剖过多则会导致将不想要的RNA包含到样品中[18,24]。
人工细胞采集/显微操作法:人工细胞采集系统由显微操纵器、倒置显微镜、微量移液管构成。可以在显微镜下观察和拍摄每个分离的单细胞,从而实现准确的分离(图1D)[19,32]。这是一种简单,方便和有效的隔离单细胞的方法,在配有膜片钳系统的电生理实验室中可以容易地进行此类显微操作。与需要从固定组织切片中分离单细胞的LCM不同,人工细胞采集对游离或培养的活细胞进行收集,显微操作在此过程中起到重要作用,这种整细胞的收集方式保证了完整的细胞分离,并最大限度地减少了在反转录步骤之前的RNA的损失[18,33]。然而,此种分选方式工作效率相对较低,同时需要操作人员具有高度的专业技能。人工细胞采集技术在分离培养的活细胞中有大量的应用,也被广泛应用于大量培养的胚胎细胞、神经元细胞中。对于数量丰富的单个游离细胞如精子细胞或培养的活细胞,可使用此方法方便的收获数量丰富的单个细胞[34-36]。
由于FACS和LCM技术的局限性,使得此两种分选技术难以在成年动物内耳组织细胞分选中加以应用。针对内耳组织的自身特点,我们将现有的人工细胞采集技术进行了改良,摸索出了适合应用于内耳组织单类细胞收集的微吸管细胞收集技术方法。
使用该方法进行内耳单类细胞收集及转录组分析具有显著优势。首先,由于内耳组织中各类细胞形态的高度特异性,使得根据细胞独特形态在显微镜下进行细胞辨认具有高度准确性,微吸管内耳细胞收集是根据细胞的独特形态来进行单种细胞收集,加之分选过程中防止污染的控制措施,可使分选出的细胞达到很高纯度。有效的避免了使用FACS分选过程中因细胞黏附所造成的杂细胞污染,以及使用LCM细胞分选过程中解剖靶细胞所带来的相邻细胞或周围其他非特异性组织的污染。
其次,这种对内耳细胞的收集方法可以避免使用FACS分选过程中对细胞的机械损伤和分选时间较长所造成的RNA的降解。通过对我们收集样本的RNA质量分析和测序过程中的质量控制指标的验证可以看出,使用微吸管收集技术获得的内耳单细胞样本的RNA质量好、纯度高,没有污染及降解,而采用FACS及LCM法均难以获得如此高质量的RNA样本。
第三,由于内耳组织的结构特点以及其他细胞分选技术所存在的技术限制,若需要对成年的哺乳动物内耳细胞样本进行单种收集,其他单细胞分选技术很难成功应用。荧光分选技术只适合于带有荧光的细胞,这大大降低了该技术的适应性。而微吸管细胞收集法的应用,可以收集到实验需要的,最能代表功能成熟的细胞转录组的成年动物的内耳细胞。
1 Fettiplace R.Hair Cell Transduction,Tuning,and Synaptic Trans⁃mission in the Mammalian Cochlea[J].Compr Physiol,2017,7(4):1197-1227.
2 Raphael Y,Altschuler RA.Structure and Innervation of the Co⁃chlea[J].Brain Res Bull,2003,60(5-6):397-422.
3 Kniss JS,Jiang L,Piotrowski T.Insights into Sensory Hair Cell Regeneration from the Zebrafish Lateral Line[J].Curr Opin Genet Dev,2016,40:32-40.
4 Géléoc GS,Holt JR.Sound Strategies for Hearing Restoration[J].Science,2014,344(6184):1241062.
5 Atkinson PJ,Huarcaya NE,Sayyid ZN,et al.Sensory Hair Cell Development and Regeneration:Similarities and Differences[J].Development,2015,142(9):1561-1571.
6 Franco B,Malgrange B.Concise Review:Regeneration in Mamma⁃lian Cochlea Hair Cells:Help from Supporting Cells Trans differen⁃tiation[J].Stem Cells,2017,35(3):551-556.
7 Hu BH.High-throughput Technologies for Gene Expression Anal⁃yses:WhatWe Have Learned for Noise-induced Cochlear Degen⁃eration?[J].JOtol,2013,8(1):25-31.
8 Burns JC,Kelly MC,Hoa M,et al.Single-cell RNA-Seq Resolves Cellular Complexity in Sensory Organs from the Neonatal Inner Ear[J].NatCommun,2015,6:8557.
9 Cai T,Jen HI,Kang H,et al.Characterization of the Transcrip⁃tome of Nascent Hair Cells and Identification of Direct Targets of the Atoh1 Transcription Factor[J].J Neurosci,2015,35(14):5870-5883.
10 Scheffer DI,Shen J,Corey DP,et al.Gene Expression by Mouse Inner Ear Hair Cells During Development[J].JNeurosci,2015,35(16),6366-6380.
11 Zheng J,ShenW,He DZ,et al.Prestin is the Motor Protein of Co⁃chlear Outer Hair Cells[J].Nature,2000,405(6783):149-155.
12 He DZ,Zheng J,Edge R,et al.Isolation of Cochlear Inner Hair Cells[J].Hear Res,2000,145(1-2),156-160.
13 Liu H,Pecka JL,Zhang Q,et al.Characterization of Transcrip⁃tomes of Cochlear Inner and Outer Hair Cells[J].J Neurosci,2014,34(33),11085-11095.
14 Li Y,Liu H,Barta CL,et al.Transcription Factors Expressed in Mouse Cochlear Inner and Outer Hair Cells[J].PLoSOne,2016,11(3):e0151291.
15 Shearer AE,Hildebrand MS,Smith RJH.Deafness and Heredi⁃tary Hearing Loss Overview.In:Adam MP,Ardinger HH,Pagon RA,et al.editors.Gene Reviews®[Internet].Seattle(WA):1993-2017.
16 Schatz DG,Swanson PC.V(D)J Recombination:Mechanisms of Initiation[J].Annu Rev Genet,2011,45,167–202.
17 Blainey PC,Quake SR.Dissecting Genomic Diversity,One Cellat a Time[J].NatMethods,2014,11(1):19–21.
18 Hodne K,Weltzien FA.Single-Cell Isolation and Gene Analysis:Pitfalls and Possibilities[J].Int J MolSci,2015,16(11):26832-26849.
19 Gross A,Schoendube J,Zimmermann S,et al.Technologies for Sin⁃gle-Cell Isolation[J].Int JMolSci,2015,16(8):16897-16919.
20 Herzenberg LA,Parks D,Sahaf B,et al.The History and Future of the Fluorescence Activated Cell Sorter and Flowcytometry:A View from Stanford[J].Clin Chem,2002,48(10):1819–1827.
21 Ibrahim S,van den Engh G.Flow Cytometry and Cell Sorting[J].Adv Biochem Eng Biotechnol,2007,106:19-39.
22 Johnson KW,Dooner M,Quesenberry PJ.Fluorescence Activated Cell Sorting:A Window on the Stem Cell[J].Curr Pharm Biote chnol,2007,8(3):133–139.
23 Basu S,Campbell HM,Dittel BN,et al.Purification of Specific Cell Population by Fluorescence Activated Cell Sorting(FACS)[J].JVis Exp,2010,10(41),pii:1546.
24 Hu P,Zhang W,Xin H,et al.Single Cell Isolation and Analysis[J].FrontCellDev Biol,2016,4:116.
25 Sugiyama T,Kim SK.Fluorescence-activated Cell Sorting Purifi⁃cation of Pancreatic Progenitor Cells[J].Diabetes Obes Metab,2008,10(Suppl4):179–185.
26 Emmert-Buck MR,Bonner RF,Smith PD,et al.LaserCaptureMi⁃crodissection[J].Science,1996,274(5289):998–1001.
27 Kummari E,Guo-Ross SX,Eells JB.Laser Capture Microdissec⁃tion–A Demonstration of the Isolation of Individual Dopamine Neurons and the Entire Ventral Tegmental Area[J].J Vis Exp,2015,96:e52336.
28 Fend F,Raあeld M.Laser Capture Microdissection in Pathology[J].JClin Pathol,2000,53(9):666–672.
29 Bonner RF,Emmert-Buck M,Cole K,et al.Laser Capture Micro⁃dissection:Molecular Analysis of Tissue[J].Science,1997,278(5342):1481-1483.
30 Espina V,Heiby M,Pierobon M,et al.Laser Capture Microdissec⁃tion Technology[J].ExpertRevMolDiagn,2007,7(5):647–657.
31 Liu A.Laser Capture Microdissection in the Tissue Biorepository[J].JBiomol Tech,2010,21(3):120–125.
32 Hodne K,Haug TM,Weltzien FA.Single-cell qPCR on Dispersed Primary Pituitary cells—An Optimized Protocol[J].BMCMol Bi⁃ol,2010,11:82.
33 Bengtsson M,Stahlberg A,Rorsman P,et al.Gene Expression Pro fi ling in Single Cells from the Pancreatic Islets of Langerhans Re⁃veals Lognormal Distribution ofmRNA Levels[J].Genome Res,2005,15(10):1388–1392.
34 Guo GJ,Huss M,Tong GQ,et al.Resolution of Cell Fate Deci⁃sions Revealed by Single-Cell Gene Expression Analysis from Zy⁃gote to Blastocyst[J].Dev Cell,2010,18(4):675–685.
35 Citri A,Pang ZP,Südhof TC,et al.Comprehensive qPCR Profi ling ofGene Expression in Single Neuronal Cells[J].NatProtoc,2011,7(1):118–127.
36 Eberwine J,Yeh H,Miyashiro K,et al.Analysis of Gene-expres⁃sion in Single Live Neurons[J].Proc Natl Acad SciUSA,1992,89(7):3010–3014.
