P-糖蛋白诱导作用的研究进展
2018-03-20陈根富阮婷婷王广基孙建国
许 悦,陈根富,熊 涛,彭 英,阮婷婷,王广基*,孙建国**
(1中国药科大学药物代谢动力学重点实验室,南京 210009;2上海药明康德新药开发有限公司药性评价部,上海 200131)
很多化合物的生物利用度(bioavailability,F)主要由促进其跨膜转运的转运体和完成体内代谢的代谢酶决定,这些化合物包括内源性物质、药物和外源性有毒异物等。转运体在保持细胞内环境稳态和排出潜在危害物质过程中起到重要作用,其中ATP结合盒(ATP-binding cassette,ABC)转运体超家族是药物转运体中最大的一类,在维持细胞生理功能中起到重要作用。其生理功能主要包括排出有害物质,摄取营养物质,转运离子、多肽和细胞信号分子等[1]。
P-糖蛋白(P-gp)是ABC转运体中最具代表性的蛋白,因其在肿瘤细胞产生多药耐药(multidrug resistance,MDR)中的作用被称为多药耐药蛋白1,编码其蛋白的基因也被称为MDR1或ABCB1。P-gp在人和其他哺乳动物的多种组织中广泛表达,尤其在肠上皮细胞的基顶侧表面、胆小管和肾近端小管表达量很高,这些器官都发挥着一定的排泄功能。外排转运体的生理功能就是转运内源性代谢产物或外源性异物,P-gp在胰腺小管、肾上腺、胎盘、睾丸、血-脑脊液屏障中也有表达[2]。
P-gp底物在大小、结构和功能上变化范围很大,包括天然产物、化疗药物、钙离子通道抑制剂、类固醇、线性或环状多肽、荧光染料等,这些化合物的化学结构、生理功能各不相同,并没有特定联系,P-gp底物的广泛性也与其对抗各种有害物质伤害的功能相一致。同时,P-gp的外排功能可被多种化合物抑制或诱导[1]。
本文结合课题组的研究,回顾了近年来有关P-gp诱导的研究进展,总结了P-gp诱导的分子机制,以及常见P-gp诱导剂的分类和特性,重点介绍了研究P-gp诱导的体外细胞模型和啮齿动物模型,以P-gp基因、蛋白表达及外排功能变化评价诱导作用强弱的实验方法。同时,本文阐述了P-gp与药物代谢酶及其他药物转运体的共同调节作用。P-gp诱导可引起多种临床药物-药物相互作用,但大量体内外研究证实,提高细胞外排功能也可作为一种临床解毒治疗的策略,如诱导P-gp表达提高可有效对抗某些P-gp底物(百草枯等)导致的细胞毒性。并且,计算机辅助设计的药效基团模型也被用于预测化合物是否为P-gp的诱导剂或激活剂。总之,P-gp诱导研究在临床前药物设计、P-gp诱导剂的筛选、潜在药物-药物相互作用预测等方面发挥一定的指导作用。
1 P-gp诱导
1.1 P-gp诱导原理
与P-gp抑制的作用效果相反,细胞通过诱导上调P-gp的表达,提高外排功能,减少外源性有害物质的伤害,从而更好地适应环境变化。短期暴露于某些外源性异物或刺激后,细胞MDR1基因被激活,一段时间后可诱导产生稳定的P-gp高表达[3]。
人MDR1基因的启动子区域是非典型的启动子序列,因其缺乏TATA盒,且含有多个启动子元件,包括:GC-box、Y-box、p53因子、反向调节因子1(invMED1)、转录激活蛋白1(AP-1)、热休克因子(HSE)、类固醇异物受体(SXR)等,这也与人P-gp转录调节的复杂性相吻合[4]。诱导MDR1基因表达的信号通路有多种,多样的基因调控网络有利于细胞快速适应外界环境的变化和对抗化学伤害。全面充分地研究MDR1基因诱导机制,有利于指导研发合适的诱导剂,用以降低有害因素造成的伤害,同时预测由诱导引起的药物相互作用。图1以利福平为例,演示了药物诱导转运体表达继而引起药物-药物相互作用的过程。

图1 药物诱导转运体表达的细胞内过程(以利福平为例)
PXR:孕甾烷X受体;RXR:视黄醛受体
1.1.1 诱导剂 P-gp可被多种药物诱导,文献报道的P-gp诱导剂种类繁多,表1列举了多种代表性的诱导剂及其分类。MDR1基因还可能被不同的细胞内因素或者环境因素诱导,如细胞因子、氧自由基、抑癌基因、热休克因子、X射线、紫外线等[1]。P-gp诱导剂的种类繁多,表明可能存在多种不同的信号通路调节MDR1的转录,使得MDR1基因能够对细胞内外环境的变化和各种刺激做出反应,维持细胞内环境稳态。
表1常见的P-gp诱导剂

药物分类P⁃gp诱导剂参考文献 抗菌药内源性物质抗肿瘤药心血管系统药抗病毒药天然产物降血脂药激素类中枢神经系统药利福平、红霉素黄体酮、醛固酮、胆红素、胆盐长春碱、道诺霉素、阿霉素、紫杉醇波生坦、安贝生坦、地高辛、维拉帕米利托那韦、安瑞那韦、茚地那韦圣约翰麦芽汁、姜黄素阿托伐他汀倍氯米松、布地奈德、地塞米松卡马西平、咖啡因、苯妥英[5-6][6-7][1,8][1,5-6,8][8-9][8,10][1,8][7,9,11][9,12]
1.1.2 P-gp诱导研究的体内外模型 药物与P-gp作用的复杂性,使得临床预测P-gp引起的药物相互作用变得更加困难,因此临床前需要合适的体外细胞模型、动物模型进行严格测试。人或动物原代细胞、组织细胞、肿瘤细胞常用于体外模拟体内环境,如用于模拟人胃肠道功能的人结肠癌细胞系Caco-2、LS180、T84、LS174T等,用于模拟血-脑脊液屏障通透性的脑微血管内皮细胞系(BMECs)等,以及用于模拟肝脏代谢的原代肝细胞、肝癌细胞等,这些细胞模型同样适用于不同组织器官P-gp的诱导研究。
研究P-gp诱导的体内模型以啮齿动物为主,主要有野生型及基因敲除的大鼠和小鼠。实验方法主要有:利用Western blot、荧光定量PCR、免疫组化等手段检测诱导前后不同组织器官(如肠道、肝、肾、血-脑脊液屏障等)P-gp基因和蛋白表达水平变化;以LC-MS/MS、放射性核素检测等方法获得特异性P-gp底物的血药浓度-时间曲线、组织分布、胃肠道吸收等参数,用以表征P-gp外排功能的变化。表2总结了有代表性的研究P-gp诱导体内外模型及实验方法。
表2P-gp诱导研究的体内外模型及实验方法

模型分类实验模型实验方法检测P⁃gp表达检测P⁃gp功能参考文献体外细胞模型人肠道上皮细胞系LS180/T84/Caco⁃2人肾脏上皮细胞系HK2人或鼠原代肝细胞人肝癌细胞HepG2人或大鼠脑微血管内皮细胞系(BMECs)WesternBlotNorthernBlot流式细胞术检测P⁃gp含量(Anti⁃P⁃gp抗体)实时荧光定量PCR(TaqMan®荧光探针)酶联免疫吸附法P⁃gp底物(罗丹明123,阿霉素,Calcein⁃AM,地高辛等)蓄积实验和外排实验P⁃gp底物细胞单层(Transwell®)双向转运实验流式细胞仪检测细胞P⁃gp底物跨膜转运[1,5,16-22]体内动物模型大鼠(血浆、肝、肾、脑、小肠等)小鼠(血浆、肝、肾、脑、小肠等)基因敲除大鼠或小鼠Westernblot检测组织中P⁃gp表达量实时荧光定量PCR检测组织中MDR1基因表达量免疫组化、在体成像诱导后P⁃gp底物药代动力学参数分析诱导后P⁃gp底物组织浓度分析制备囊膜外排、摄取实验在体肠灌流、原位脑灌注[1,23-29]
1.2 P-gp和药物代谢酶的共同调节
P-gp与药物代谢酶CYP3A4的诱导存在共同调节机制。据统计,CYP3A4参与了超过半数已上市药的代谢,因此二者的共同诱导对于药物的吸收、分布、代谢、排泄(ADME)有着重要影响[2,13-14]。很多P-gp底物同时是CYP3A4的底物,P-gp和CYP3A4不仅在底物特异性上有大量重合,并且存在一定的互补表达模式,这种表达模式在肝和肠壁中尤为重要。因此,同时通过CYP3A家族代谢和P-gp转运的药物,其口服生物利用度受诱导作用的影响更大,存在药物-药物相互作用的可能性大。
Sérée等[11]研究发现,地塞米松可同时诱导HepG2细胞中MDR1和CYP3A4基因表达,在小鼠肝脏中mdr1b和cyp3a基因也存在同样的现象。多种核受体可能是参与MDR1和CYP3A4共同诱导的重要因素,例如孕甾烷X受体(PXR)、组成型雄甾烷受体(CAR)等。Synold等[15]发现,紫杉醇可通过PXR介导的信号通路同时提高肝细胞中P-gp和CYP3A4的蛋白表达。
药物代谢酶和外排转运体的共同调节,有利于细胞的解毒和排泄,使机体能够对抗多种外来有害异物的侵害。CYP3A4和P-gp的共同诱导作用可能限制或改变很多药物的生物利用度,研究二者的共同诱导对指导药物发现和临床安全合理用药有着重要意义。此外,阐明CYP3A4和P-gp的共同诱导机制,寻找双诱导剂,可能是一种临床解毒治疗的新途径。
1.3 P-gp和其他转运体的共同调节
大量研究表明,除P-gp以外的其他ABC转运体也可被诱导,并且有多种转录因子参与其中,如PXR参与诱导MRP1,CAR、PXR参与诱导MRP2和MRP3[30]。在外周血单核细胞(PBMC)中MDR1、MRP2、BCRP的mRNA表达水平与PXR表达呈正相关[31]。Narang等[32]发现地塞米松能提高血-脑脊液屏障中多种转运体的表达和功能。大鼠原代脑微血管内皮细胞(RBMEC)给予地塞米松后,Bcrp、Mdr1和Mrp2基因表达显著上调,其蛋白表达和功能亦显著提高,且这种诱导呈剂量依赖性。随着给药停止,该诱导作用是可逆的。Cermanova等[33]报道,大鼠多次经口给予胺碘酮25 mg/(kg·d),其肾P-gp和MRP2表达显著提高,且P-gp底物罗丹明123和内源性物质结合胆红素在肾的清除显著增加。大量体内外实验数据表明,某些药物可同时诱导多种转运体的表达,提示在人体内,转运体诱导可能降低药物的生物利用度或影响其分布,产生药物-药物相互作用,造成疗效下降甚至治疗失败。
1.4 P-gp激活
化合物与P-gp快速作用而提高其外排功能的方式被称为P-gp的激活。与诱导不同,激活的过程并不涉及P-gp蛋白表达量变化。P-gp诱导剂通过提高P-gp的表达量增强细胞外排能力,而P-gp激活剂则直接与P-gp结合,使其发生构象变化,促使P-gp底物结合到其他转运位点上。因此,P-gp的激活不涉及基因转录、蛋白翻译过程,作用速度比诱导更加快速。有学者利用两种P-gp荧光底物罗丹明123和Hoechst-33342证明了P-gp存在多个底物结合位点的猜测[34]。
近年来,P-gp高分辨晶体结构的发现表明:其底物结合区不是独立的、不连续的药物结合位点,而是一个很大的、可变性很强的区域[35]。该区域含有大量的亲水性供电子和受电子基团、带电荷基团、含苯环氨基酸等,由此形成大量有利于药物结合的亚结构。这些可变性强的底物结合区域可与多种药物发生亲脂性相互作用、氢键结合、静电相互作用等,进而造成空间结构的改变,影响其与底物的结合。P-gp的激活有利于细胞更为快速地应对外界环境变化,更迅速地排出有害物质,维持内环境稳态。
1.5 P-gp诱导引起的临床药物-药物相互作用
胃肠道中P-gp主要位于肠细胞的肠腔侧,可将底物泵回肠腔内,且表达量从小肠的近端到远端呈递增趋势。因此,P-gp的外排作用限制了其底物的口服生物利用度。另外,P-gp参与肠腔的主动分泌,可影响静脉注射P-gp底物的系统清除率。由于P-gp底物结构类型变化范围大,诱导和抑制是P-gp引起药物-药物相互作用的主要因素。胃肠道P-gp诱导可能导致其底物(地高辛、他林洛尔等)的口服生物利用度下降、系统清除率上升。中枢神经系统(CNS)中血-脑脊液屏障有大量P-gp表达,疾病和药物诱导P-gp表达和功能的升高,可能限制CNS药物穿透血-脑脊液屏障,降低治疗效果。表3列举了部分因P-gp诱导产生的临床药物-药物相互作用。
2 P-gp诱导作为治疗策略的体内外研究
P-gp在近十几年来一直被视为肿瘤细胞多药耐药的重要因素,对其抑制的研究取得大量成果。如前文所述,P-gp在肝、肾、脑、胃肠道、前列腺等正常组织中也有表达,其外排底物的广泛性有利于机体细胞对抗有害物质。因此,可利用P-gp诱导有效地降低细胞内有害物质蓄积,进而起到解毒作用,这种假设在以百草枯(paraquat,PQ)为模型药物的实验体系中被大量证实[1,18,42-44]。
百草枯是一种含氯高效除草剂,也是敏感的P-gp底物。误服或摄入百草枯可能导致帕金森病(Parkinson′s disease),严重的甚至导致死亡,并且尚无有效的解毒方法。有报道称在动物体内通过诱导P-gp可对百草枯中毒进行解救[44],实验中大鼠给予25 mg/kg百草枯2 h后,单剂量给予P-gp诱导剂地塞米松100 mg/kg,大鼠肺部P-gp表达显著提高,并且肺中百草枯的蓄积量下降40%,大鼠的存活率上升了50%。同时,给予诱导剂还显著提高了大鼠肠道和肝脏中P-gp的表达量,降低了百草枯在肠道中吸收。在其他体外细胞模型中,诱导P-gp对抗百草枯中毒的作用也有报道。Silva等[43]研究发现,阿霉素和金丝桃素(圣约翰麦芽汁发挥诱导作用的主要活性成分之一)均能显著提高Caco-2细胞中P-gp的表达和功能,并且这种提高呈时间和浓度依赖性。由百草枯引起的细胞死亡率随着两种诱导剂的加入显著下降,而加入特异性P-gp抑制剂能显著降低诱导剂的解毒效果,进一步证明这种解毒作用确实是由于P-gp表达上调所引起,提示临床可采用诱导转运体为策略进行中毒的解救。
表3P-gp诱导引起的临床药物-药物相互作用
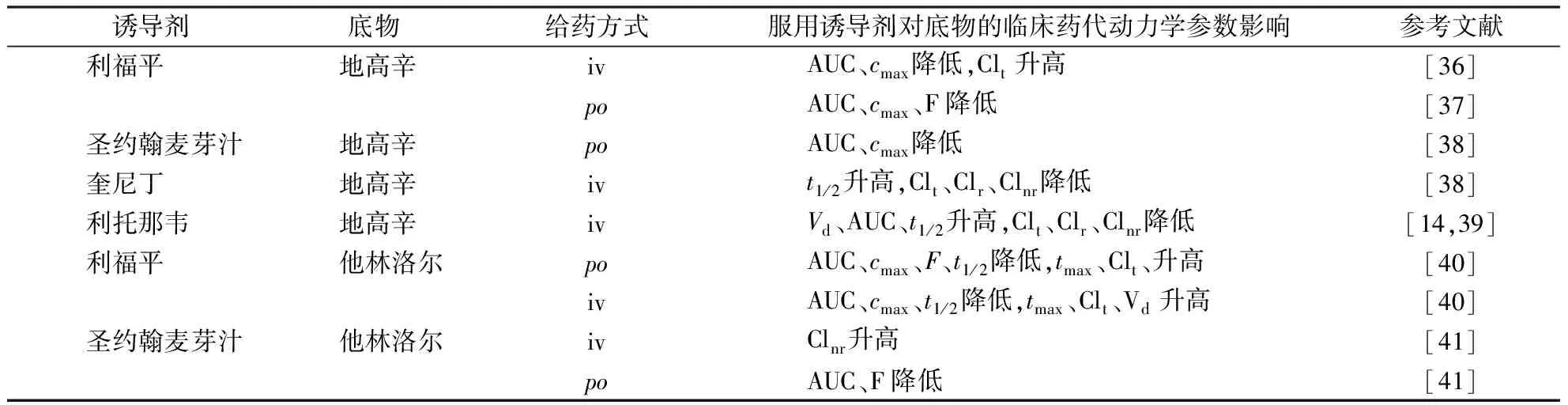
诱导剂底物给药方式服用诱导剂对底物的临床药代动力学参数影响参考文献利福平地高辛ivAUC、cmax降低,Clt升高[36]poAUC、cmax、F降低[37]圣约翰麦芽汁地高辛poAUC、cmax降低[38]奎尼丁地高辛ivt1/2升高,Clt、Clr、Clnr降低[38]利托那韦地高辛ivVd、AUC、t1/2升高,Clt、Clr、Clnr降低[14,39]利福平他林洛尔poAUC、cmax、F、t1/2降低,tmax、Clt、升高[40]ivAUC、cmax、t1/2降低,tmax、Clt、Vd升高[40]圣约翰麦芽汁他林洛尔ivClnr升高[41]poAUC、F降低[41]
3 计算机辅助研究P-gp诱导
利用细胞或动物实验研究新化学实体(new chemical entities,NCEs)和P-gp的相互作用关系,费时费力且价格昂贵。因此计算机辅助模型开始应用于P-gp底物、抑制剂的预测,此类模型大多通过定量构效关系分析(QSAR analyses)、药效基团模型、分子对接技术等手段进行预测[19,45-46]。然而,现阶段研究新型P-gp诱导剂和激动剂主要方式是随机筛选,工作量较大,且准确性较差。为了解决这一难题,Silva等[22]基于文献报道,以34种P-gp诱导剂建立一种P-gp诱导药效基团模型。该模型的工作原理如图2所示:首先对多种P-gp诱导剂按性质进行无重叠的划分,对其配体聚类分析后输出配体群集,接着组合构建多个药效基团。构建好的模型用4种已知的P-gp诱导剂(安瑞那韦、奈非那韦、嘌呤霉素和育亨宾)进行验证后,可用于预测新化学实体是否为P-gp诱导剂或激活剂。

图2 P-gp诱导药效基团模型工作流程
该药效基团模型主要通过文献报道的诱导剂或激活剂建立,但由于这些化合物结构差异巨大,用一个通用的药效基团预测所有化合物显然不合适,所以要先进行配体聚类分析,并用已知的诱导剂进行模型验证。作者将该模型用于新合成的氧杂蒽酮(xanthones)、硫杂蒽酮(thioxanthones)衍生物诱导活性的筛选,发现计算机预测结果与体外细胞实验数据一致性良好,证明该P-gp诱导药效基团模型可快速有效地预测化合物诱导活性[42]。
4 新合成化合物的筛选
从新药开发的角度而言,早期进行化合物的药代动力学特性筛选对后期药物研发的成功率有着重要影响。因此,近年来更多的研究开始关注新化学实体是否是P-gp的诱导剂或者激活剂[19,47]。
氧杂蒽酮和硫杂蒽酮是药物化学中十分重要的化合物,因为其母核有抗肿瘤等多种显著生物活性,是很有潜力的药物前体。Silva等[43]用Caco-2细胞进行硫杂蒽酮衍生物的筛选,发现数种有P-gp诱导活性的衍生物。利福平是常用的P-gp诱导剂,Vilas-Boas等[48]通过筛选发现一种利福平衍生物RedRif能够显著提高RBE4细胞P-gp外排功能。细胞给予10 mol/L RedRif孵育24 h后,P-gp功能显著增强;孵育48 h后,P-gp蛋白表达显著提高。在百草枯细胞毒性实验中,RedRif可有效对抗百草枯引起的细胞毒性。
大量体外实验表明,可以使用某些细胞模型比较化合物对P-gp诱导能力的强弱,进而为研究其体内相互作用提供一定的数据支持。业界也在探索准确高效评价P-gp诱导特性的体外细胞模型、体内动物模型和计算机辅助药物设计方法。本课题组分别从P-gp外排功能、基因表达和蛋白表达水平比较了3种人结肠癌细胞系Caco-2、LS180、T84作为P-gp诱导体外模型的优劣。发现LS180细胞诱导潜力较好,对多种常见的P-gp诱导剂反应较为敏感,是较好的研究肠道转运体诱导特性的体外模型,并且可以实现高通量筛选。另一种细胞T84在Transwell®双向转运系统中细胞单层完整性良好,且其多种代谢酶、转运体的表达量与人体肠道细胞相接近[49-50]。本课题组利用T84细胞测试30余种已上市药物的细胞渗透性,发现表观渗透系数与人体口服吸收数据相关性较好,与经典的药物渗透性评价细胞模型Caco-2、MDR1-MDCK数据一致性良好,提示T84细胞Transwell®双向转运体系是较好的研究口服药物渗透性的模型(本部分数据待发表)。
5 结 语
美国FDA在《药物相互作用研究指导原则》中建议:当试验药物为P-gp底物时,应研究试验药物的转运是否会被抑制或诱导,可选用P-gp抑制剂(如利托那韦、环孢素或维拉帕米)或诱导剂(如利福平)进行研究[51]。P-gp的诱导可以有效提高细胞外排作用,降低有毒P-gp底物在细胞内蓄积而产生的毒性。各种体内体外实验证明了诱导剂可提高细胞P-gp的表达和外排功能,验证了诱导P-gp产生解毒作用的有效性,提示这一策略可用于临床解毒治疗。总之,P-gp诱导研究将为新药的临床前评价以及临床安全合理用药提供有力的数据支持,对诱导引起的药物-药物相互作用做出合理解释,对新药研发提供一定的指导。
[1] Silva R,Vilas-Boas V,Carmo H,etal.Modulation of P-glycoprotein efflux pump:induction and activation as a therapeutic strategy[J].PharmacolTher,2015,149:1-123.
[2] Schinkel AH.P-Glycoprotein,a gatekeeper in the blood-brain barrier[J].AdvDrugDelivRev,1999,36(2/3):179-186.
[3] Zhuo W,Hu L,Lv J,etal.Role of pregnane X receptor in chemotherapeutic treatment[J].CancerChemotherPharmacol,2014,74(2):217-227.
[4] Hennessy M.A primer on themechanics of P-glycoprotein the multidrug transporter[J].PharmacolRes,2007,55(1):1-15.
[5] Weiss J,Theile D,Spalwisz A,etal.Influence of sildenafil and tadalafil on the enzyme- and transporter-inducing effects of bosentan and ambrisentan in LS180 cells[J].BiochemPharmacol,2013,85(2):265-273.
[6] Naruhashi K,Kurahashi Y,Fujita Y,etal.Comparison of the expression and function of ATP binding cassette transporters in Caco-2 and T84 cells on stimulation by selected endogenous compounds and xenobiotics[J].DrugMetabPharmacok,2011,26(2):145-153.
[7] Manceau S,Giraud C,Declèves X,etal.ABC drug transporter and nuclear receptor expression in human cytotrophoblasts:influence of spontaneous syncytialization and induction by glucocorticoids[J].Placenta,2012,33(11):927-932.
[8] Haslam IS,Jones K,Coleman T,etal.Induction of P-glycoprotein expression and function in human intestinal epithelial cells (T84)[J].BiochemPharmacol,2008,76(7):850-861.
[9] Abuznait AH,Kaddoumi A.Role of ABC transporters in the pathogenesis of Alzheimer′s disease[J].ACSChemNeurosci,2012,3(11):820-831.
[10] He J,Tang J,Yang WH,etal.P-gp induction by curcumin:an effective antidotal pathway[J].JBioequivBioavail, 2013,5(6):236-241.
[11] Sérée E,Villard PH,Hevér A.Modulation of MDR1 and CYP3A expression by dexamethasone:evidence for an inverse regulation in adrenals[J].BiochemBiophysResCommun,1998,252(2):392-395.
[12] Wen T,Liu YC,Yang HW,etal.Effect of 21-day exposure of phenobarbital,carbamazepine and phenytoin on P-glycoprotein expression and activity in the rat brain[J].JNeurolSci,2008,271(1):99-106.
[13] Foti RS.“Target-Site” drug metabolism and transport[J].DrugMetabDispos,2015,43(8):1156-1168.
[14] Müller F,Fromm MF.Transporter-mediated drug-drug interactions[J].Pharmacogenomics,2011,12(7):7-21.
[15] Synold TW,Dussault I,Forman BM.The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux[J].NatMed,2001,7(5):584-590.
[16] Liu L,Collier AC,Link JM,etal.Modulation of P-glycoprotein at the human blood-brain barrier by quinidine or rifampin treatment:a positron emission tomography imaging study[J].DrugMetabDispos,2015,43(11):1795-1804.
[17] Sager JE,Yu J,Ragueneau-Majlessi I,etal.Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches:a systematic review of published models,applications,and model verification[J].DrugMetabDispos,2015,43(11):1823-1837.
[18] Silva R,Carmo H,Dinis-Oliveira R,etal.Invitrostudy of P-glycoprotein induction as an antidotal pathway to prevent cytotoxicity in Caco-2 cells[J].ArchToxicol,2011,85(4):315-326.
[19] Silva R,Carmo H,Vilas-Boas V,etal.Colchicine effect on P-glycoprotein expression and activity:insilicoandinvitrostudies[J].ChemBiolInteract,2014,218:50-62.
[20] Zerin T,Kim YS,Hong SY,etal.Protective effect of methylprednisolone on paraquat-induced A549 cell cytotoxicity via induction of efflux transporter,P-glycoprotein expression[J].ToxicolLett,2012,208(2):101-107.
[21] Silva R.Pglycoprotein induction in Caco 2 cells by newly synthetized thioxanthones prevents paraquat cytotoxicity[J].ArchToxicol2015,89:18.
[22] Silva R,Sousa E,Carmo H,etal.Induction and activation of P-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the P-glycoprotein substrate paraquat[J].ArchToxicol,2014,88(4):937-951.
[23] Fenner KS,Troutman MD,Kempshall S,etal.Drug-drug interactions mediated through P-glycoprotein:clinical relevance andinvitro-invivocorrelation using digoxin as a probe drug[J].ClinPharmacolTher,2009,85(2):173-181.
[24] Reitman ML,Chu X,Cai X,etal.Rifampin′s acute inhibitory and chronic inductive drug interactions:experimental and model-based approaches to drug-drug interaction trial design[J].ClinPharmacolTher,2011,89(2):234-242.
[25] Fromm MF.Importance of P-glycoprotein at blood-tissue barriers[J].TrendsPharmacolSci,2004,25(8):423-429.
[26] Schwarz UI,Hanso H,Oertel R,etal.Induction of intestinal P-glycoprotein by St John′s wort reduces the oral bioavailability of talinolol[J].ClinPharmacolTher,2007,81(5):669-678.
[27] Liu J,Zhou F,Chen Q,etal.Chronic inflammation up-regulates P-gp in peripheral mononuclear blood cells via the STAT3/Nf-kappab pathway in 2,4,6-trinitrobenzene sulfonic acid-induced colitis mice[J].SciRep,2015,5:13558.
[28] Wen T,Liu YC,Yang HW,etal.Effect of 21-day exposure of phenobarbital,carbamazepine and phenytoin on P-glycoprotein expression and activity in the rat brain[J].JNeurolSci,2008,270(1):99-106.
[29] Jin S,Yao D,Liu C,etal.Effect of chronic liver failure on the function and expression of P-GP and MRP2 in rat brain[J].JChinaPharmUniv(中国药科大学学报),2012,43(1):64-69.
[30] Callaghan R,Luk F,Bebawy M.Inhibition of the multidrug resistance P-glycoprotein:time for a change of strategy[J]?DrugMetabDispos,2014,42(4):623-631.
[31] Albermann N,Schmitz-Winnenthal FH,Z′graggen K,etal.Expression of the drug transporters MDR1/ABCB1,MRP1/ABCC1,MRP2/ABCC2,BCRP/ABCG2,and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver[J].BiochemPharmacol,2005,70(6):949-958.
[32] Narang VS,Fraga C,Kumar N.Dexamethasone increases expression and activity of multidrug resistance transporters at the rat blood-brain barrier[J].AmJPhysiolCellPhysiol,2008,295(2):440-450.
[33] Cermanova J,Fuksa L,Brcakova E,etal.Up-regulation of renal Mdr1 and Mrp2 transporters during amiodarone pretreatment in rats[J].PharmacolRes,2010,61(2):129-135.
[34] Safa AR.Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators[J].CurrMedChemAnticancerAgents,2004,4(1):1-17.
[35] Aller SG,Yu J,Ward A.Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding[J].Science,2009,323(5922):1718-1722.
[36] Drescher S,Glaeser H,Mürdter T,etal.P-glycoprotein mediated intestinal and biliary digoxin transport in humans[J].ClinPharmacolTher,2003,73(3):223-231.
[37] Greiner B,Eichelbaum M,Fritz P,etal.The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin[J].JClinInvest,1999,104(2):147-153.
[38] Gurley BJ,Swain A,Williams DK,etal.Gauging the clinical significance of P-glycoprotein-mediated herb-drug interactions:comparative effects of St.John′s wort,Echinacea,clarithromycin,and rifampin on digoxin pharmacokinetics[J].MolNutrFoodRes,2008,52(7):772-779.
[39] Montanari F,Ecker GF.Prediction of drug-ABC-transporter interaction-Recent advances and future challenges[J].AdvDrugDelivRev,2015,86:17-26.
[40] Westphal K,Weinbrenner A,Zschiesche M,etal.Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings:a new type of drug-drug interaction[J].ClinPharmacolTher,2000,68(4):345-355.
[41] Schwarz UI,Hanso H,Oertel R.Induction of intestinal P-glycoprotein by St John′s wort reduces the oral bioavailability of talinolol[J].ClinPharmacolTher,2007,81(5):669-678.
[42] Silva R.Induction and activation of P-glycoprotein by dihydroxylated xanthones protect against the cytotoxicity of the P-glycoprotein[J].ArchToxicol,2014,88(4):15.
[43] Silva R,Palmeira A,Carmo H,etal.P-glycoprotein induction in Caco-2 cells by newly synthetized thioxanthones prevents paraquat cytotoxicity[J].ArchToxicol,2015,89(10):1783-1800.
[44] Dinis-Oliveira RJ,Remiao F,Duarte JA,etal.P-glycoprotein induction:an antidotal pathway for paraquat-induced lung toxicity[J].FreeRadicBiolMed,2006,41(8):1213-1224.
[45] Murgueitio MS,Bermudez M,Mortier J,etal.Insilicovirtual screening approaches for anti-viral drug discovery[J].DrugDiscoveryToday:Technologies,2012,9(3):e219-e225.
[46] Lu JR,Wang FX,Li YM,etal.Synthesis,biological evaluation and 2D-QSAR study of a series of isoflavone derivatives as modulators of multidrug resistance[J].JChinaPharmUniv(中国药科大学学报),2013,44(4):296-302.
[47] Padala AK,Wani A,Vishwakarma RA,etal.Functional induction of P-glycoprotein efflux pump by phenyl benzenesulfonamides:synthesis and biological evaluation of T0901317 analogs[J].EurJMedChem,2016,122:744-755.
[48] Vilas-Boas V,Silva R,Palmeira A.Development of novel rifampicin-derived P-glycoprotein activators/inducers.Synthesis,in silico analysis and application in the RBE4 cell model,using paraquat as substrate[J].PLoSOne,2013,8(8):e74425.
[49] Yamaura Y,Chapron BD,Wang Z,etal.Functional comparison of human colonic carcinoma cell lines and primary small intestinal epithelial cells for investigations of intestinal drug permeability and first-pass metabolism[J].DrugMetabDispos,2016,44(3):329-335.
[50] Bourgine J,Billaut-Laden I,Happillon M,etal.Gene expression profiling of systems involved in the metabolism and the disposition of xenobiotics:comparison between human intestinal biopsy samples and colon cell lines[J].DrugMetabDispos,2012,40(4):694-705.
[51] FDA.Guidance for industry,drug interaction studies-study design,data analysis,and implications for dosing and labeling recommendations[S].2012.
