儿童Gitelman综合征3例临床特点及基因分析
2018-01-03徐爱晶李秀珍郑锐丹
徐爱晶 苏 玲 李秀珍 程 静 郑锐丹
广州妇女儿童医疗中心遗传与内分泌科 (广东广州 510623)
儿童Gitelman综合征3例临床特点及基因分析
徐爱晶 苏 玲 李秀珍 程 静 郑锐丹
广州妇女儿童医疗中心遗传与内分泌科 (广东广州 510623)
目的 探讨儿童Gitelman综合征的临床及基因突变特点。方法 回顾分析3例Gitelman综合征患儿的临床资料。结果 3例患儿均为男性,年龄分别为3、8、10岁。临床表现为低钾血症、低镁血症、碱中毒、高肾素血症、高醛固酮血症。基因检测显示存在SLC12A3基因的复合杂合突变,共发现SLC12A3基因的5个突变位点:c.179C>T(Thr60Met)、c.248 G>A(Arg83Gln)、c.2129 C>A(Ser710X)、c.2660+1G>A、c.1456G>A(Asp486Asn)。患儿确诊后,经补钾、补镁、螺内酯治疗后病情好转。结论 儿童出现低钾血症需注意Gitelman综合征,基因检测有助于明确诊断。
Gitelman综合征; SLCl2A3基因; 临床特征; 基因突变
Gitelman综合征(Gitelman syndrome,GS,OMIM 263800)是一种常染色体隐性遗传病,主要是由编码肾脏远曲小管钠-氯协同转运蛋白(Na+-Clcotransporter,NCCT)的SLC12A3基因突变造成,临床表现为低血钾、低氯性代谢性碱中毒、低血镁、低尿钙、继发性醛固酮增多以及血压正常或偏低。GS起病较隐匿,常在青春期或成年后发病,症状不严重,儿童期起病报道较少,容易漏诊、误诊。本研究回顾分析2015年1月-2016年1月在广州妇女儿童医疗中心住院、经SLC12A3基因检测确诊Gitelman综合征3例患儿的临床资料。
1 临床资料
例1,男,3岁6个月,因反复低血钾2年余就诊。患儿于1岁4个月因手足口病在当地医院住院期间发现血钾2.1 mmol/L,予补钾后血钾正常。其后每年4、5次因呼吸道感染、急性胃肠炎住院治疗,均发现血钾低下,予补钾后正常。本次入院前2周因咳嗽、发热,拟诊细菌性肺炎在当地医院住院,查血钾1.61 mmol/L,血钙0.90 mmol/L,予补钾、补钙、抗感染及对症治疗后,咳嗽好转,发热消退,但血钾仍低。患儿自发病以来无乏力、四肢瘫痪、多饮、多尿等不适。家族中无类似疾病史。患儿系G1P1,足月剖宫产,出生体质量3.3 kg,无出生窒息史,母妊娠史无特殊。入住广州妇女儿童医疗中心体格检查:体温36.5 ℃,心率132次/min,呼吸22次/min,收缩压(SBP)/舒张压(DBP)95/58 mmHg;体质量13 kg,身长92 cm;行为发育大致正常;无特殊体征。
例2,男,8岁9个月,因发作性手麻3年,发作后不能走路2个月,抽搐1次入院。患儿于入院前3年起,天冷时出现双侧或单侧无名指发麻,伴疼痛,持续数分钟至数小时,无发热、头痛、头晕、恶心、呕吐,无抽搐发作、意识不清,予按摩或保暖后可好转,每年发作3~5次,当地诊所治疗无明显好转。其后发作时渐出现双手指屈曲,持续数分钟至数小时,每月2、3次,至多家医院多次查血钾偏低,波动在1.64~2.88 mmol/L。头颅磁共振成像(MRI)未见异常。双手发麻时予补钾对症治疗症状可以缓解,未规律服药及定时复查血钾。入院前16天患儿无明显诱因出现抽搐1次,表现为双手握拳、双眼上翻、口吐白沫、面色发绀、四肢抖动、呼之不应,持续约半小时,经当地医院静脉用药(具体不详)后痉止。查头颅MRI、颅脑CT血管造影(CTA)未见明显异常。脑电图示异常脑电图(清醒/睡眠)间歇期,左半球慢波增多。多次查血钾波动在1.61~2.24 mmol/L,考虑“癫痫,低钾血症”转至广州妇女儿童医疗中心就诊。家族中无类似疾病史。患儿系G1P1,足月顺产,出生体质量2.75 kg,无窒息史,母妊娠史无特殊。入院体格检查:体温36.6 ℃,心率96次/min,呼吸20次/min,SBP/DBP108/58 mmHg,体质量18 kg,身高115.7 cm;行为发育无异常;无特殊体征。
例3,男,10 岁2个月,因反复低血钾2年余入院。入院前2年患儿因头晕伴乏力外院检查示低血钾,自述病情好转后出院,其后未服药治疗。入院前1个月因发热、呕吐去当地医院就诊,查血钾2.54 mmol/L,血钠139.0 mmol/L,血氯95.0 mmol/L,予对症处理后病情好转;入院前5天再次因乏力住院,住院期间反复出现低血钾,最低至2.52 mmol/L,予补钾等对症处理后恢复正常。患儿自发病来,无头晕、心悸、胸闷,无寒战、抽搐,无多饮多尿,无恶心、呕吐、腹胀,无烦躁不安,精神、胃纳及睡眠可。家族中无类似疾病史。患儿系G5P2,足月剖宫产,出生体质量3.0 kg,无窒息史,母妊娠史无特殊。入住广州妇女儿童医疗中心体格检查:体温36.2 ℃,心率88次/min,呼吸22次/min,SBP/DBP102/78 mmHg,体质量28 kg,身高136.2 cm;行为发育无异常;无特殊体征。
3例患儿的血常规、尿常规、空腹血糖、肝肾功能检查均未见异常;血气、电解质检查示低血钾代谢性碱中毒、低血镁、低尿钙;甲状腺功能、甲状旁腺功能、皮质醇未见异常;肾素、醛固酮水平均增高(表1)。例1患儿胰岛素样生长因子(IGF)-1低于同年龄儿童的最低水平。3例患儿均行双肾B超检查未见明显异常。
在获得患儿及家属知情同意后,采集患儿及其父母外周血2 mL,提取基因组DNA,采用PCR产物直接测序法对SLC12A3基因(NC_000016)26个外显子及其相邻内含子进行突变分析。同时对CLCNKB基因进行DNA测序。3例患儿的突变位点通过直接测序分析,共发现SLCl2A3基因的6个突变位点,其中5个突变位点可能与GS相关,其中包括3个错义突变、1个无义突变、1个剪切突变。例1的7号外显子的突变位点未见报道,7号外显子的突变位点意义未明。见表2、图1。
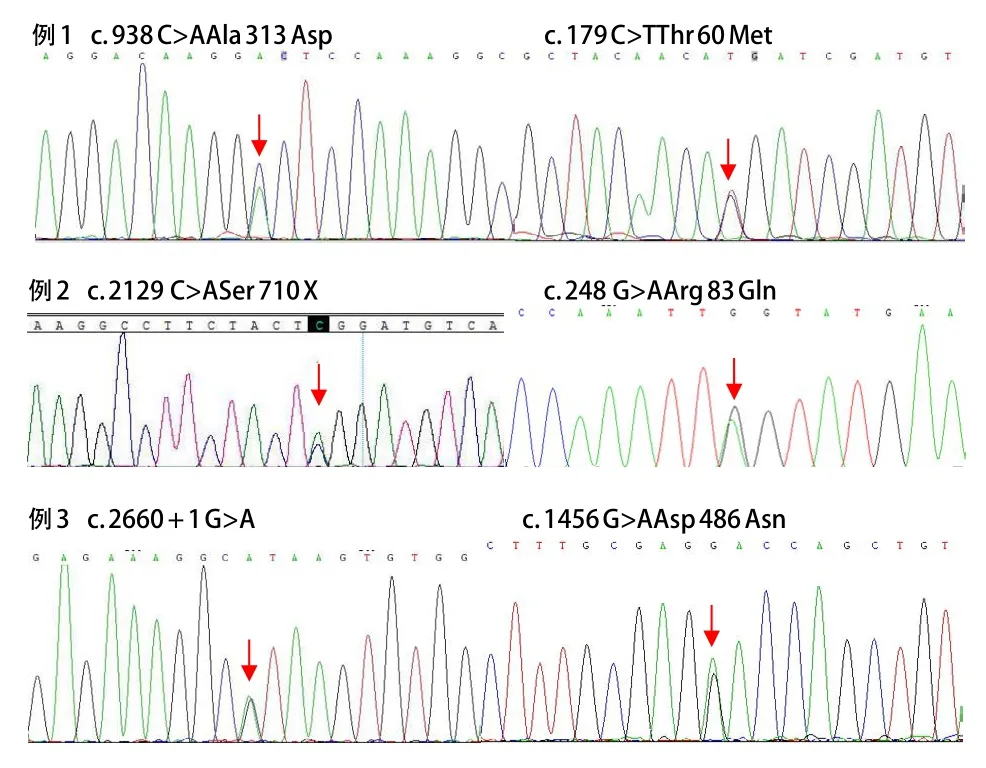
图1 3例患儿SLCl2A3基因测序结果
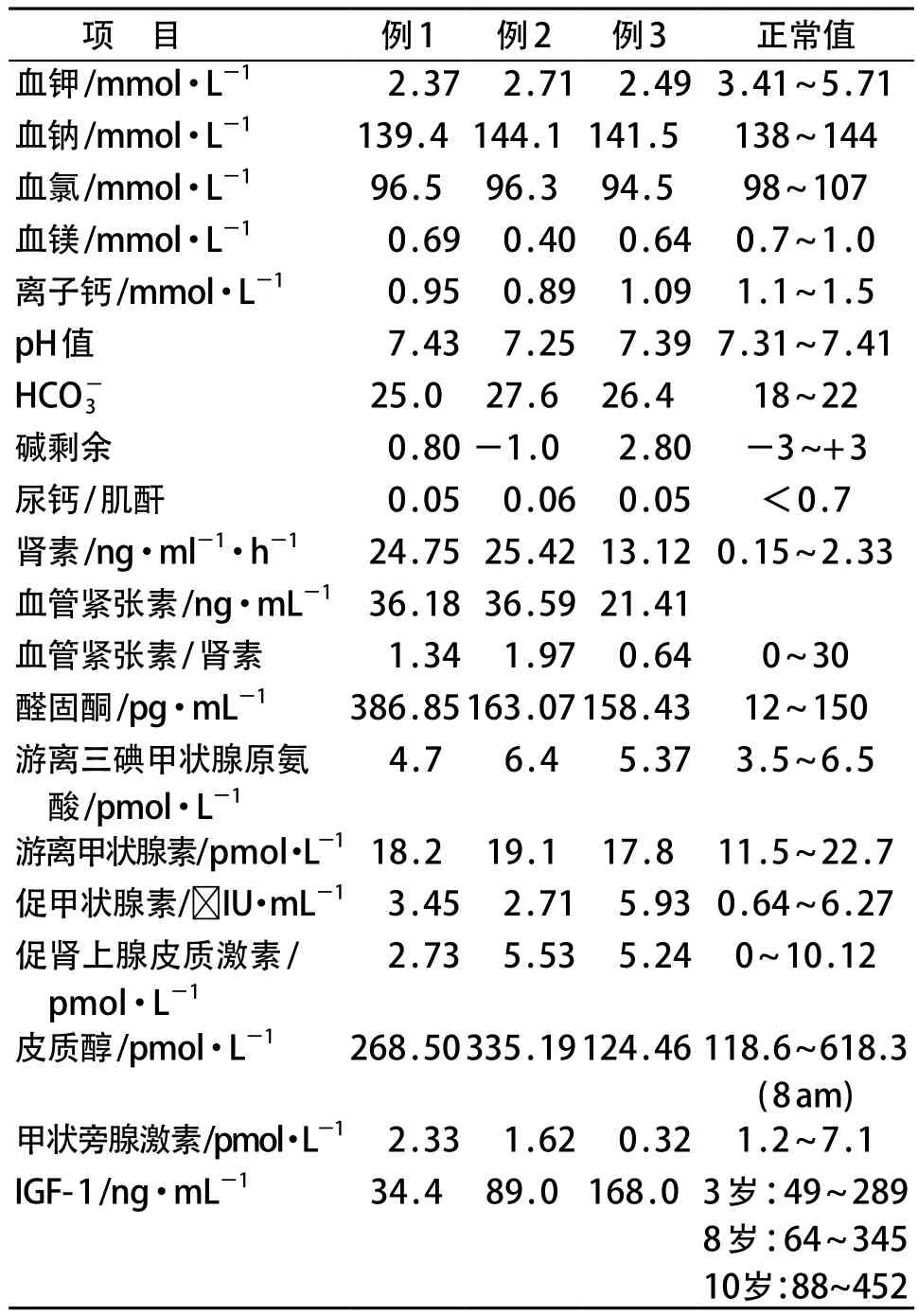
表1 患儿实验室检查结果

表2 患儿基因突变位点
3例患儿经确诊后均给予补钾治疗,以10%氯化钾口服液、10%硫酸镁及螺内酯治疗,出院前复查血钾分别为3.6 mmol/L、3.73 mmol/L、3.45 mmol/L,血镁分别为0.73 mmol/L、0.71 mmol/L、0.82 mmol/L。
2 讨论
GS在欧洲发病率约为1/40 000,日本为10.3/10 000[1],既往认为中国发病率较低,关于GS的报道多为散在的病例,儿童发病的GS报道则更少。曾经认为GS是Bartter综合征的一个亚型,但是随着对疾病遗传学和分子机制研究的深入,目前已经明确这两种疾病具有不同发病机制和遗传学背景。1966年Gitelman等报道了3例临床表现与Bartter综合征相似,但同时合并有低镁血症和低尿钙的成年女性患者。1996年Simon等[2,3]使用基因连锁分析对12个不相关的家系进行分析,发现位于染色体长臂16q13的SLCl2A3基因是GS发病的致病基因。该基因编码噻嗪类利尿剂敏感的NCCT,NCCT由12个跨膜结构域组成,该结构域的羧基端和氨基端都在细胞内。NCCT主要表达在肾脏远曲小管,是包含1 021个多肽氨基酸的蛋白质,从肾小球滤过的Na+和Cl-有5%~10%在此重吸收。SLAC12A3基因突变损害肾小管正常的Na+/Cl-联合转运功能,导致肾性失盐性疾病,表现为低血钾、低血镁、代谢性碱中毒、低尿钙,肾素-血管紧张素系统(renin-angiotensin system,RAS)激活,血压正常或偏低。GS早期症状通常不明显,表现有乏力、虚弱、口渴、夜间遗尿等,影响生活质量。因为症状不明显,早期诊断比较困难。
一般认为Barrter综合征可见于任意年龄段,但多发于5岁前儿童,而GS的症状常发生于6岁之后,但大部分诊断时间为青少年和成年期[4]。GS发病初期表现通常是偶然发现的无症状和孤立的低钾血症。部分患儿表现为疲劳、头晕、肌肉无力、痉挛、呕吐、腹痛、发热、夜尿增多及多尿,偶有低血压出现。严重低钾血症和低镁血症可以影响生长发育。本研究通过对临床表现为代谢性碱中毒、低钾血症、低镁血症的患儿进行SLAC12A3基因检查,发现3例患儿起病年龄在3.6~10岁,从起病到确诊时间平均为2.5年,表明本病在儿童期间并不罕见。儿童期症状不典型,加之对本病的认识不足,常常认为低钾血症是由于呕吐、腹泻所致而未进一步追查病因,易导致诊断延误。目前GS的临床诊断基于以下标准:低钾血症,代谢性碱中毒,高尿钾(>25 mmol/24 h),低镁血症(<0.66 mmol),低尿钙/肌酐之比(<0.2),肾素-血管紧张素系统活性增加但血压正常。但需要除外代谢性低钾血症,胃肠丢失钾,肾小管酸中毒以及长期服用缓泻剂、利尿剂及药物成瘾等[5]。
有研究认为,性别与GS临床症状的严重程度相关,男性患者的病情重于女性。欧洲GS男性患者病情重于女性,主要表现为发病年龄更早,青少年时的生长、发育延迟等。SLC12A3基因突变的性质/位置和性别可能共同决定了GS严重程度[6]。Lin等[7]也发现,男性患者较女性出现更低的血钾水平、更严重的症状和更早的发病年龄。陈楠等[8]研究显示,男性GS患者的夜尿发生要多于女性,肾脏受累的GS患者均为男性。性别因素对表型的影响可能与雌激素有关。本组3例均为男性,是否是因为男性病情相对较重而容易被及早发现有关,尚需进一步研究。
SLC12A3基因位于染色体16q13,大小约55 kb,包括26个外显子。目前文献中已报道SLC12A3基因突变超过250种,突变类型包括无义突变、错义突变、移码突变、缺失、插入和剪切位点突变,其中75%以上为错义突变,没有明显的突变热点。本研究3例患儿共发现SLC12A3基因5个突变位点可能是GS致病性突变,包括3个错义突变、1个无义突变、1个剪切突变,其中2个位于氨基端,1个位于羧基端,4个位于钠-氯共转运子胞浆段(包括胞浆内氨基端和羧基端的突变)。经检索人类基因突变库(HGMD)和最近文献,例1中c.938 C>A为新突变意义不明,经生物信息预测软件预测致病可能性小。虽然只有1个突变位点,但例1患儿临床表现典型。根据既往文献报道有20%~40%左右的GS患儿仅发现1个突变位点[9-11]。中国人群中发现T60M和D486N是常见的突变位点[12,13],本组例1患儿携带T60M突变位点,例3患儿携带D486N。在众多突变位点中,错义突变T60M突变位点为亚洲人群高发突变位点。最近研究认为Thr60在激活NCC的关键磷酸化位点上起重要作用,NCC携带突变体 p.T60M几乎完全丧失其内在活性却并不影响其表面表达[14],并且其突变显著抑制了Thr46和Thr55的磷酸化,破坏了NCC蛋白的内在活性[15]。D486N是另一常见的突变位点,邵乐平等[12]报道一组67例GS患者SLC12A3基因突变41种,其中D486N突变率为13.4%,居于频发突变第2位。另一项对42例患者研究发现,52个突变位点中D486N突变率为19.7%,为频发突变第1位[13]。目前没有发现SLC12A3基因型与表现型有明确的关系,有文献提示血镁水平与病情的严重程度相关[16]。
通常情况下,GS患儿生长发育不受影响,但当存在严重的低钾血症和低镁血症可能导致生长发育迟缓[17],经补钾、补镁治疗后自然生长速度并没有改善。动物实验显示,给予大鼠低钾饮食可导致身高、体质量增长缓慢,同时生长激素及IGF-1水平降低,表明低钾对生长激素的分泌起负性调节作用[18],并且低钾可以导致生长激素、IGF-1组织特异性的改变。多项研究报道指出,GS综合征和Bartter综合征可以合并生长激素完全或部分缺乏[19,20]。Ko等[20]报道1例9岁10个月男童,GS综合征合并矮身材,生长激素激发试验示生长激素完全缺乏(胰岛素、左旋多巴及可乐定激发试验,生长激素峰值为3.2 ng/mL),经予生长激素治疗后生长速度12 cm/年,停用生长激素后生长速度降低为3.6 cm/年,并且低镁血症经生长激素治疗后好转。另一项包含4例GS患者的报道,生长激素治疗第一年生长速度10~12 cm/年,同样低镁血症改善[21]。动物实验表明,应用生长激素能明显增加镁的吸收和保留[22]。本组3例患儿中2例存在生长发育迟缓,该2例患儿IGF-1水平低于同年龄段水平或位于正常值低限。在儿童GS患儿出现明显的生长迟缓,需要检测GH-IGF-1轴的功能,在纠正低钾血症后仍不能获得满意的身高增长速度,需要给予GH治疗。
对于无症状的GS患儿,可以不予药物治疗,但需要每年监测电解质1、2次,给予高钠、高钾饮食。低镁血症患者需要终生服用镁制剂,因应用大剂量镁制剂容易出现腹泻,所以较难保持血镁水平正常。口服氯化镁起始剂量可以4~5mg/kg,分4、5次服用,以免引起腹泻。根据血镁水平调整剂量。当出现感染、呕吐、腹泻等症状时,应该增加镁量。当出现抽搐发作时可静脉予20% MgCl2,0.1 mmol/(kg·次),必要时可隔6小时重复使用[23]。出现低血钾时可补充大量的氯化钾,氯化钾1~3 mmol/(kg·d),分3、4次,儿童可达10 mmol/kg,成人500 mmol/d,但需要注意胃的耐受性。非选择性醛固酮受体拮抗剂螺内酯有抑制醛固酮合成、拮抗醛固酮的作用,可以减少肾脏钾离子的排泄和丢失。选择性醛固醇阻滞剂依普利酮(eplernone)也适用于治疗GS,但临床应用尚缺乏大样本的数据支持。肾脏远曲小管Na+-Cl-转运阻滞剂——阿米洛利可以通过选择性地抑制上皮细胞NaCl通道从而减少醛固酮敏感的Na+-Cl-交换,也可用于治疗GS。阿米洛利[5~10mg/(1.73m2·d)]治疗应以较低剂量开始以避免出现低血压。若出现软骨钙质沉着病(伪痛风发作)需要使用非甾体类消炎药(NSAID)[24]。
总之,儿童期发现的不明原因的低钾血症,需考虑到Gitelman综合征,早期诊断,早期治疗,提高患儿的生活质量,并积极进行基因诊断和遗传咨询。
[1] Tago N, Kokubo Y, Inamoto N, et al. A high prevalence of Gitelman's syndrome mutations in Japanese [J]. Hypertens Res, 2004, 27(5): 327-331.
[2] Mastroianni N, De Fusco M, Zollo M, et al. Molecular cloning, expression pattern, and chromosomal localization of the human Na-Cl thiazide-sensitive cotransporter (SLC12A3)[J]. Genomics, 1996, 35(3): 486-493.
[3] Mastroianni N, Bettinelli A, Bianchetti M, et al. Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome [J]. Am J Hum Genet,1996, 59 (5): 1019-1026.
[4] Cruz DN, Shaer AJ, Bia MJ, et al. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life [J]. Kidney Int, 2001, 59(2): 710-717.
[5] Sinha A, Lněnička P, Basu B, et al. Gitelman syndrome: novel mutation and long-term follow-up [J]. Clin Exp Nephrol,2012, 16(2): 306-309.
[6] Vargas-Poussou R, Dahan K, Kahila D, et al. Spectrum of mutations in Gitelman syndrome [J]. J Am Soc Nephrol, 2011, 22(4): 693-703.
[7] Lin SH, Cheng NL, Hsu YJ, et al. Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/cloride cotransporter [J]. Am J Kidney Dis, 2004, 43(2): 304-312.
[8] 秦岭, 邵乐平, 任红, 等. Gitelman综合征的表型特点及性别因素对表型的影响 [J]. 中华肾脏病杂志, 2009, 25(7)∶532-537.
[9] Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters [J]. Physiol Rev,2005, 85(2): 423-493.
[10] Nakhoul F, Nakhoul N, Dorman E, et al. Gitelman's syndrome: a pathophysiological and clinical update [J].Endocrine, 2012, 41(1): 53-57.
[11] Riveira-Munoz E, Chang Q, Bindels RJ, et al. Gitelman's syndrome: towards genotype-phenotype correlations? [J].Pediatr Nephrol, 2007, 22(3): 326-332.
[12] 邵乐平, 逯静茹, 郎艳华, 等. 中国Gitelman综合征患者的基因型、表型分析及随访研究 [J]. 中华内分泌代谢杂志 , 2017, 33(1)∶ 40-46.
[13] Wang F, Shi C, Cui Y, et al. Mutation prof i le and treatment of Gitelman syndrome in Chinese patients [J]. Clin Exp Nephrol, 2017, 21(2): 293-299.
[14] Miao Z, Gao Y, Bindels RJ, et al. Coexistence of normotensive primary aldosteronism in two patients with Gitelman's syndrome and novel thiazide-sensitive Na-Cl cotransporter mutations [J]. Eur J Endocrinol, 2009, 161(2): 275-283.
[15] Richardson C, Raf i qi FH, Karlsson HK, et al. Activation of the thiazide-sensitive Na+-Cl-cotransporter by the WNK-regulated kinases SPAK and OSR1 [J]. J Cell Sci, 2008,121(Pt 5): 675-684.
[16] Jiang L, Chen C, Yuan T, et al. Clinical severity of Gitelman syndrome determined by serum magnesium [J]. Am J Nephrol, 2014, 39(4): 357-366.
[17] Riveira-Munoz E, Chang Q, Godefroid N, et al.Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome [J]. J Am Soc Nephrol, 2007, 18(4): 1271-1283.
[18] Gil-Peña H, Garcia-Lopez E, Alvarez-Garcia O, et al.Alterations of growth plate and abnormal insulin-like growth factor I metabolism in growth-retarded hypokalemic rats:effect of growth hormone treatment [J]. Am J Physiol Renal Physiol, 2009, 297(3): F639-F645.
[19] Buyukcelik M, Keskin M, Kilic BD, et al. Bartter syndrome and growth hormone deficiency∶ three cases [J]. Pediatr Nephrol, 2012, 27(11)∶ 2145-2148.
[20] Ko CW, Koo JH. Recombinant human growth hormone and Gitelman's syndrome [J]. Am J Kidney Dis, 1999, 33(4): 778-781.
[21] Slyper AH. Growth, growth hormone testing and response to growth hormone treatment in Gitelman syndrome [J]. J Pediatr Endocrinol Metab, 2007, 20(2): 257-259.
[22] Min SR, Cho HS, Hong J, et al. Gitelman syndrome combined with complete growth hormone def i ciency [J]. Ann Pediatr Endocrinol Metab, 2013, 18(1): 36-39.
[23] AI Shibli A, Narchi H. Bartter and Gitelman syndromes: spec trum of clinical manifestations caused by different mutations[J]. World J Methodol, 2015, 5(2): 55-61.
[24] Knoers NV, Levtchenko EN. Gitelman syndrome [J].Orphanet J Rare Dis, 2008, 3: 22.
Clinical characteristics and gene analysis in three children with Gitelman syndrome
XU Aijing, SU Ling, LI Xiuzhen,CHENG Jing, ZHENG Ruidan (Department of Pediatric Endocrinology and Metabolism, Guangzhou Women and Children's Medical Center, Guangzhou Medical University, Guangzhou 510623, Guangdong ,China)
Objectives To explore the clinical and gene mutation characteristics of Gitelman syndrome in children.Method The clinical data of 3 children with Gitelman syndrome were retrospectively analyzed. Results All three cases were male and their age were 3, 8 and 10 years . The clinical manifestations were hypokalemia, hypomagnesemia, alkalosis,hyperreninemia, and hyperaldosteronemia. Gene detection revealed a complex heterozygous mutation in the SLC12A3 gene. A total of 5 mutation sites were found in the SLC12A3 gene, c.179C>T (Thr60Met), c.248 G>A (Arg83Gln), c.2129 C>A (Ser710X),c.2660+1G>A, c.1456G>A (Asp486Asn). After the diagnosis was confirmed, they were treated with potassium supplement,magnesium supplement, and spironolactone and the conditions were improved in all cases. Conclusions In children with hypokalemia, be aware of Gitelman syndrome, and gene detection is helpful for the diagnosis.
Gitelman syndrome; SLCl2A3 gene; clinical manifestations; gene mutation
doi∶10.3969/j.issn.1000-3606.2017.12.003
广州市医药卫生科技一般引导项目(No.20141A011028)
徐爱晶 电子信箱∶ xuaj246@126.com
2017-03-31)
蔡虹蔚)
