SHOC2基因突变致Noonan综合征1例报告
2018-01-03梅玉霞常国营王秀敏
梅玉霞 常国营 庄 承 丁 宇 李 娟 李 辛 王 剑 王秀敏
1.上海中医药大学附属上海市第七人民医院(上海 200137);2.上海交通大学医学院附属上海儿童医学中心(上海 200127)
SHOC2基因突变致Noonan综合征1例报告
梅玉霞1常国营2庄 承1丁 宇2李 娟2李 辛2王 剑2王秀敏2
1.上海中医药大学附属上海市第七人民医院(上海 200137);2.上海交通大学医学院附属上海儿童医学中心(上海 200127)
目的 探讨SHOC2基因突变导致Noonan综合征(NS)的临床表型及分子诊断。方法 回顾分析1例NS患儿的临床资料及基因检测结果。结果 患儿,男,8个月。自出生后即存在喂养、睡眠困难,易哭吵,生长缓慢发育落后。头围偏大,头发稀疏、细黄,前额宽大突出,鼻梁扁平,眼距略宽,双侧眼裂向外下略倾斜,无眼睑下垂。彩色多普勒超声心动图显示卵圆孔未闭,室间隔与左室稍肥厚。在患儿SHOC2基因中找到“新生突变(De novo)”,杂合错义变异c.4A>G,p.Ser2Gly,其父母此位点为正常基因型。经查阅相关文献资料发现,睡眠困难这一临床表现目前在SHOC2基因突变类型NS患者中尚无类似报道。结论 SHOC2基因突变所致NS,其临床表型跟国外报道基本一致。睡眠困难可能是SHOC2基因突变型NS的一个新的表型谱。
Noonan综合征; SHOC2基因; 突变; 临床表型
Noonan综合征(Noonan syndrome,NS)于1968年由 Jacqueline Noonan 首次报道,是一类常染色体显性遗传性疾病,具有完全的外显率但表达可变,累及多个器官系统。主要临床特征表现为特殊面容、先天性心脏病、身材矮小、发育迟缓、肾脏泌尿系统畸形等。NS在活产儿中的发病率约为1∶2 500~1∶1 000[1,2]。2001年前 NS主要依据其特征性临床表型来诊断,随着分子诊断技术的发展和运用,发现约70%的NS患者存在基因突变,目前已明确与NS相关的基因有PTPN11、SOS1、KRAS、NRAS、RAF1、BRAF、SHOC2、CBL、MAP2K1、A2ML1、SOS2和RIT1[3,4]。现回顾1例NS患者的临床表型、实验室检查、基因检测结果,并结合查阅复习相关文献资料,以期提高临床医师对NS表型和基因型的认识,希望为临床诊断、治疗和遗传咨询提供更好的参考依据。
1 临床资料
迟缓伴喂养困难8个月于2016年9月来上海儿童医学中心就诊。患儿系G2P2,母孕期常规产检,无妊娠合并症和并发症,胎儿检查未发现明显异常,孕37+6周顺产娩出(臀位产);出生体质量3.3 kg,身高50 cm,出生时无窒息抢救史,Apgar评分具体不详。生后母乳喂养,但吃奶量少,喂奶时出现吸吮无力、吞咽困难、容易呛奶,1月龄时体质量为3.1 kg。3月龄体质量为3.5 kg,因吸吮困难由母乳喂养改配方奶喂养,但进食量少,每次约15~30 mL。此外,患儿出生后数日开始出现反复哭吵(不分昼夜),睡眠困难,主要表现为睡眠时间短暂,每次睡眠时间约10~30 min,24 h累计睡眠时间约3~4 h。患儿大小便无异常,体温无异常,无明显气促、青紫、呼吸困难等。患儿家族中无类似疾病史,父母非近亲结婚,均体健,无遗传性疾病史,有一哥哥2岁,生长发育无异常。
入院体格检查:身高62 cm(<P3),体质量5.2 kg(<P3)。神志清,发育落后(8月龄能翻身、抬头,不能独坐,不会呀呀发音),听力无异常,易激惹,呼吸平稳,面色、反应可,全身浅表淋巴结未触及,未见咖啡斑及色素沉着;头围47 cm,头发稀疏、细黄,前额宽大突出,鼻梁扁平,眼距略宽,双侧眼裂向外下略倾斜,无眼睑下垂,无眼球震颤,双侧瞳孔等大等圆,对光反射灵敏;鼻唇沟对称,牙齿未萌出,高腭弓,无腭裂,咽部略红;能抬头,颈短,后发际线低,双侧耳朵对称,耳位偏后偏低;胸廓无明显畸形,乳距宽,两肺呼吸音清,未闻及罗音;心律齐,心前区未闻及病理性杂音;腹部软,肝脾肋下未触及,四肢肌张力低下,下肢明显,肌力3~4级,双侧膝反射、跟腱反射无异常;男性外阴,阴囊小,皮褶少,睾丸能降入阴囊,大小约1 mL;布氏征(-),克氏征(-)。患儿外部主要表现见图1。
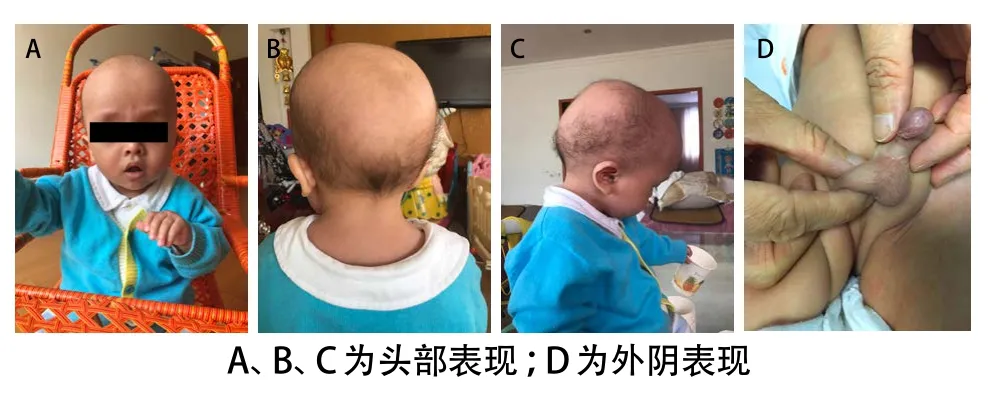
图1 Noonan综合征患儿主要临床特征
实验室检查:血常规、C反应蛋白(CRP)、降钙素原、尿常规、肝肾功能、出凝血时间、甲状腺功能、血尿串联质谱、肌酸激酶(CK)、肌酸激酶同工酶(CKMB)、免疫球蛋白、电解质、脑脊液检查均正常,甲状旁腺素(PTH)0.31 pmol/L偏低。脑电图提示轻度异常。头颅MRI平扫示双侧脑室偏大,双侧额颞部蛛网膜下隙增宽,双侧脑室后角旁白质信号减少,枕大池区蛛网膜囊肿。头颅CT示双侧侧脑室稍大,枕大池稍大。彩色多普勒超声心动图示卵圆孔未闭,室间隔与左室稍肥厚,左心收缩功能正常范围。心电图示窦性心律,电轴左偏,T波变化。胸椎正侧位片示第7胸椎椎体前缘似呈子弹头样改变,余胸椎未见异常。
经家属知情同意后,分别采集患儿及父母的外周血2 mL提取DNA,采用全外显子测序方法分析目的基因。再利用Sanger测序验证经二代测序分析的SHOC2基因的突变。使用UCSC在线软件(http ∶//genome.ucsc. edu /index. html)设计SHOC2(GenBank序列号:NM_007373.3)基因变异位点的扩增引物。使用rTaqDNA聚合酶(日本TaKaRa公司)经行聚合酶链反应(PCR),PCR产物经电泳后使用QIAquick Gel Extraction Kit(德国Qiagen公司)进行纯化,纯化后产物经ABI3730XL测序仪(美国Applied Biosystems公司)Sanger测序,测序数据使用美国SoftGenetics公司Mutation Surveyor软件进行分析。
基因检测结果显示,在患儿SHOC2基因中找到杂合的错义变异c.4A>G,p.Ser2Gly。Sanger测序验证家系成员,发现其父母此位点为正常基因型,因此该变异为“新生突变(De novo)”。见图2。

图2 Noonan综合征患儿及其父母基因检测结果
患儿在随访过程中,于 9个月时因“生长发育迟缓,发热6天”入住上海市儿童医学中心神经内科,经头孢呋辛抗感染,氨基酸肌醇维生素B12,脾氨肽口服冻干粉提高免疫力,氯硝基安定帮助睡眠,高热卡深度水解配方鼻饲喂养等治疗后出院。出院后仍在内分泌科、神经内科、营养科、心血管科定期随访至今。共发生呼吸道感染2次,当地医院儿科门诊抗炎后病愈。因“腹泻、呕吐”在当地医院儿科就诊2次,对症治疗后症状消失。最近一次随访时患儿1岁4个月,仍易激惹、烦躁,睡眠时间较前略有改善,每次1~2 h,24 h累计睡眠时间6~7 h,奶量500~600 mL/d,添加部分半流质辅食,50~100 g/d。能有意识说1~2个字,能独坐,扶站片刻,不能抬腿、行走。体质量7.41 kg(< P3),身高68.5 cm(< P3),头围48 cm(P50~ P75),头发稀疏、脆,面部特征同前,乳距宽,胸廓无明显畸形,肋软骨轻度外翻,心肺无异常,四肢肌张力无明显低下,肌力3~4级。彩色多普勒超声心动图示卵圆孔已闭合,室间隔及左室壁稍肥厚,左心收缩功能正常。头颅MRI示胼胝体中后1/3略偏薄,小脑扁桃体位置稍低,枕大池扩张,双侧眼距宽。甲状腺功能无异常,心肌酶无异常,胰岛素样生长因子(IGF-1)<25 ng/mL(51~303 ng/mL),IGF-1结合蛋白3(IGF-BP3)1.6 μg/mL(0.8~3.9 μg/mL)。
2 讨论
目前认为,丝裂原活化蛋白酶信号转导通路(RAS-MAPK)的信号上调是导致NS发病的根本原因。在该通路中编码激活TRKs、RAS蛋白,RAS功能的调节器,以及下游信号传感器的基因出现突变会导致RAS相关性疾病。NS是最常见的RAS相关疾病[5,6]。其主要特征包括身材矮小,先天性心脏缺陷,异常的骨骼畸形,典型的面部特征如前额宽阔、睑裂高度偏斜、向下倾斜 ,眼睑下垂、眼距宽,耳朵向下向后方旋转成角等。目前为止,共报道有12个基因与NS以及类NS样疾病的发病有关,分别是PTPN11、SOS1、KRAS、NRAS、RAF1、BRAF、SHOC2、CBL MAP2K1、A2ML1、SOS2和 RIT1基因。
2003年Mazzanti等[7]发现一类面部特征类似NS,同时伴有生后生长缓慢、轻度智力障碍、头发稀疏易落、先天性心脏缺陷、皮肤色素沉着、色素痣等表现的患者,当时缺乏分子生物学诊断依据,称之为合并有头发稀疏的Noonan样综合征(Noonan-like syndrome with loose anagen hair,NS/LAH)。后来,研究者对25例NS/LAH患者进行分子生物学检查,证实其生物学基础为SHOC2基因的c.4A>G,p.Ser2Gly错义变异[8]。
SHOC2基因具有7个外显子,位于染色体10q25上,可以被广泛表达,它的c.4A>G,p.Ser2Gly突变可以提供1个十四酰化氨基末端的识别位点,十四饱和脂肪酸可以结合该位点,导致目标基因表达从而上调RAS-MAPK通路信号。具体突变基因的表达临床有其相应的表型,SHOC2基因c.4A>G,p.Ser2Gly错义突变的NS患者主要表现为具有典型NS特殊面部特征、身材矮小、认知障碍、明显多动行为[8,9]。此外,还表现为头发异常、心脏缺陷、皮肤色素沉着、喂养困难、骨骼异常、头围增大、掌纹细多深等。研究报道,约80%患者有肌张力减低和巨颅,60%存在羊水过多、巨大儿和喂养困难[10]。头发异常是区别于其他NS最突出的表型,包括头发细、脆容易扯断、稀疏、生长缓慢。目前报道的大部分患者都存在头发异常,仅极少数患者无此特征,可能跟患者死于婴儿早期,头发表现不典型有关[11]。文献检索未发现国内报道由SHOC2基因突变引起的NS病例,本例患儿的临床表型同国外报道基本一致,主要表现有NS婴儿期特殊的面部特征,出生后明显的喂养困难、生长迟缓,发育落后,头发稀疏易脱落,头围大,外生殖器阴囊皱褶少,肌张力肌力低下。结合临床表型并利用二代测序排除相关基因后,从患者SHOC2基因中找到杂合的错义变异c.4A>G,p.Ser2Gly。目前报道,约80%以上NS患者合并有心脏病变,其中以肺动脉狭窄最常见,占40%~50%,肥厚性心肌病占10%~25%[12,13]。大部分NS/LAH患者同样存在心脏病变,但主要表现为二尖瓣异常和室间隔缺损,个别报道有快速进展的肥厚性心肌病[11]。本例患儿彩色多普勒超声心动图显示存在先天性卵圆孔未闭合。除以上常见表型外,少数患者合并恶性肿瘤[9,10]。尽管目前尚无证据表明,SHOC2基因突变与人类肿瘤发病相关,但相关病例报道表明,在NS/LAH患者中SHOC2基因突变增加某些特异性肿瘤的患病风险,如神经母细胞瘤、横纹肌肉瘤、脑瘤、急性白血病等[14]。本例患儿尽管从目前的相关检查中未发现存在恶性肿瘤的依据,但仍需要密切随访观察。
除上述特征性表现外,本例患儿还存在明显的睡眠困难,这一表型在国外已报道的SHOC2基因突变的NS病例中未被描述过。有研究表明,SHOC2基因参与调节神经细胞分化过程,当SHOC2基因损耗时可以导致神经生长因子减少从而影响神经轴突的生长发育[15]。因此,本例患儿出现明显的睡眠困难可能与SHOC2基因突变导致神经系统生长发育异常有关。目前在已有的病例报道中尚无此表型描述,仍需增加样本数据进一步证实。
关于NS的治疗,目前以对症治疗为主,建议多学科联合随访。先天性心脏异常如肺动脉狭窄患者可根据狭窄不同程度,选择定期随访、介入治疗或外科手术治疗。存在严重喂养困难者要营养科定期随访,根据喂养困难程度和营养状况调整喂养方案。在NS患者中,很多患者下丘脑-生长激素-胰岛素样生长因子轴存在异常,表现为生长激素缺乏症(GHD)或对生长激素(GH)不敏感[16]。SHOC2基因突变的NS多伴有明显的生长发育迟缓,NS/LAH患者普遍存在GH缺乏和/或低水平的IGF-1和IGF-BP3[17]。早在2007年,美国食品药品监督局就推荐伴有身材矮小的NS
患者可予重组人生长因子(rhGH)治疗。尽管对于长期应用rhGH治疗的效果和安全性仍存在不确定性,
但很多研究表明rhGH治疗可提高IGF-1的水平、加快生长速率,改善NS患者的成年身高,而且在密切随访过程中未发现明显不良反应[16,18,19]。甚至有学者利用rhGH治疗NS/LAH不仅可以改善患者身高还可以改善肌力和肌张力[20]。
[1] Noonan JA. Hypertelorism with Turner phenotype. A new syndrome with associated congenital heart disease [J] . Am J Dis Child, 1968, 116(4)∶ 373-380.
[2] Roberts AE, Allanson JE, Tartaglia M, et al. Noonan syndrome[J]. Lancet, 2013, 381(9863)∶ 333-342.
[3] Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome [J]. Am J Hum Genet, 2013, 93 (1)∶ 173-180.
[4] Aoki Y, Niihori T, Inoue S, et al. Recent advances in RASopathies [J]. J Hum Genet, 2016, 61 (1)∶33-39.
[5] Cirstea IC, Kutsche K, Dvorsky R, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome [J]. Nat Genet,2010, 42(1)∶ 27-29.
[6] Martinelli S, De Luca A, Stellacci E, et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome like phenotype [J]. Am J Hum Genet, 2010,87(2)∶ 250-257.
[7] Mazzanti L, Cacciari E, Cicognani A, et al. Noonan-like syndrome with loose anagen hair∶ a new syndrome? [J]. Am J Med Genet A, 2003, 118A(3)∶ 279-286.
[8] Cordeddu V, DiSchiavi E, Pennacchio LA, et al. Mutation of SHOC2 promotes aberrant protein Nmyristoylation and causes Noonan-like syndrome with loose anagen hair [J]. Nat Genet,2009, 41(9)∶ 1022-1026.
[9] Ekvall S, Hagenas L, Allanson J, et al. Co-occurring SHOC2 and PTPN11 mutations in a patient with severe/complex Noonan syndrome-like phenotype [J]. Am J Med Genet A,2011, 155A(6)∶ 1217-1224.
[10] Karen W, Dina J, Laurie D, et al. Expanding the SHOC2 mutation associated phenotype of noonan syndrome with loose anagen hair∶ structural brain anomalies and myelofibrosis [J].Am J Med Genet A, 2013, 161A(10)∶ 2420-2430.
[11] Hoban R, Roberts AE, Demmer L, et al. Noonan syndrome due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant [J].Am J Med Genet A, 2012, 158A(6)∶ 1411-1413.
[12] Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome∶ clinical features, diagnosis, and management guidelines [J]. Pediatrics, 2010, 126(4)∶ 746-759.
[13] Zmolikova M, Puchmajerova A, Hecht P, et al. Coarctation of the aorta in Noonan-like syndrome with loose anagen hair [J].Am J Med Genet A, 2014, 164A(5)∶ 1218-1221.
[14] Garavelli L, Cordeddu V, Errico S, et al. Noonan syndromelike disorder with loose anagen hair∶ A second case with neuroblastoma [J]. Am J Med Genet A, 2015, 167A(8)∶ 1902-1907.
[15] Leon G, Sanchez-Ruiloba L, Perez Rodriguez A, et al. Shoc2/Sur8 protein regulates neurite out growth [J]. PLoS One, 2015,9(12)∶ e114837.
[16] Mazzanti L, Tamburrino F, Scarano E, et al. GH Therapy and first final height data in Noonan-like syndrome with loose anagen hair (Mazzanti syndrome) [J]. Am J Med Genet A,2013, 161A(11)∶ 2756-2761.
[17] Lo FS, Wang CJ, Wong MC, et al. Moyamoya disease in two patients with Noonan-like syndrome with loose anagen hair [J].Am J Med Genet Part A, 2015, 167(6)∶ 1285-1288.
[18] S,☒klar Z, Genens M, Poyrazog︶lu S,, et al. The growth characteristics of patients with noonan syndrome∶ results of three years of growth hormone treatment∶ a nationwide multicenter study [J]. J Clin Res Pediatr Endocrinol, 2016,8(3)∶ 305-312.
[19] Jeonq I, Kang E, Cho JH, et al. Long-term efficacy of recombinant human growth hormone therapy in shortstatured patients with Noonan syndrome [J].Ann Pediatr Endocrinol Metab, 2016, 21(1)∶26-30.
[20] Takasawa K, Takishima S, Morioka C, et al. Improved growth velocity of a patient with Noonan-like syndrome with loose anagen hair (NS/LAH) without growth hormone deficiency by low-dose growth hormone therapy [J]. Am J Med Genet A,2015, 167A(10)∶ 2425-2429.
Noonan syndrome caused by mutation of SHOC2 gene: a case report
MEI Yuxia1, CHANG Guoying2, ZHUANG Cheng1, DING Yu2, LI Juan2, LI Xin2, WANG Jian2, WANG Xiumin2(1.The Seventh People’s Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai 200137, China; 2.Shanghai Children’s Medical Center, Shanghai Jiaotong University School of Medicine, Shanghai 200127, China)
Objective To investigate the clinical phenotype and molecular diagnosis of Noonan syndrome (NS) caused by mutations in SHOC2 gene. Methods The clinical data and gene testing results of one child with NS were analyzed retrospectively. Results This is an 8-month-old infant. Since birth, the boy had feeding and sleeping diff i culties, irritability,and growth retardation. The boy had large head circumference, sparse, thin and yellow hair, broad and prominent forehead, fl at nose, slightly wide eye distance, and slightly bilateral eye fi ssure outward tilt, no eyelid ptosis. Echocardiography showed patent foramen ovale, ventricular septum and left ventricular hypertrophy. A novel mutation (De novo) was found in the SHOC2 gene,heterozygous missense mutation c.4A>G, p.Ser2Gly His parents were normal genotypes. According to the clinical characteristics,relevant literature was reviewed. The clinical manifestation of sleep difficulty has not been reported in the NS patients with SHOC2 mutation. Conclusions This is the fi rst domestic reported NS case with SHOC2 mutation. The phenotype is consistent with the foreign reports. Sleep diff i culty may be a new phenotype of NS with SHOC2 mutation.
Noonan syndrome; SHOC2 gene; mutation; clinical phenotype
doi∶10.3969/j.issn.1000-3606.2017.12.006
王秀敏 通信作者:wangxiumin1019@126.com
患儿,男,8个月,汉族,浙江台州人,因生长发育
2017-06-28)
邹 强)
