儿童种痘水疱病样淋巴组织增生性疾病临床病理分析
2017-11-20傅思莹李秋燕匡忠生谢宇晖
傅思莹,齐 庆,李秋燕,匡忠生,谢宇晖
儿童种痘水疱病样淋巴组织增生性疾病临床病理分析
傅思莹1,齐 庆2,李秋燕2,匡忠生1,谢宇晖1
目的探讨儿童种痘水疱病样淋巴组织增生性疾病的临床病理学特征、诊断、鉴别诊断及预后。方法收集6例儿童种痘水疱病样淋巴组织增生性疾病,观察其临床表现、组织病理学、免疫表型及相关分子病理检测,收集随访资料并复习相关文献。结果男童4例,女童2例,病史1个月~4年。临床均表现为反复发作的水痘样疱疹并伴发热。镜下见表皮内有水疱形成或伴坏死,真皮层至皮下脂肪层内有数量多少不等的淋巴样细胞浸润,细胞可为轻至中度异型,多位于小血管及附属器周围。免疫组化示异型细胞可表达CD2、CD3、CD5、CD7、CD43、CD4、CD8、TIA-1,仅1例表达CD56,所有病例均不表达CD20、Pax-5。Ki-67增殖指数平均42.3%。5例EBER原位杂交检测呈阳性,2例TCR克隆性重排阳性。5例获得随访,1例患儿死亡。结论儿童种痘水疱病样淋巴组织增生性疾病临床较为少见,与慢性活动性EB病毒感染密切相关,临床过程可能为独立的疾病谱系,其性质可为良性、交界性及恶性。病理诊断需密切结合临床表现。
种痘水疱病;淋巴瘤;Epstein-Barr病毒;临床病理特征
种痘水疱病样淋巴组织增生性疾病是发生于皮肤与EB病毒(Epstein-Barr virus, EBV)感染相关的疾病,临床表现类似种痘水疱病(hydroa vacciniforme, HV),但其病变范围更广,无法自行消退,并有可能转化为种痘水疱病样淋巴瘤(hydroa vacciniforme-like lymphoma, HVLL)。WHO(2008)淋巴造血系统肿瘤分类中EBV阳性T细胞淋巴组织增殖性疾病包括:儿童系统性EBV阳性T细胞淋巴增殖性疾病和HVLL[1];新版WHO(2006)淋巴组织肿瘤分类将HVLL更名为HV样淋巴组织增生性疾病[2]。该病临床较少见,病理诊断较为困难。本文现报道6例HV样淋巴组织增生性疾病,观察其临床及病理学特征,探讨其诊断及鉴别诊断。
1 材料与方法
1.1材料收集2010年1月~2016年12月确诊的HV样淋巴组织增生性疾病6例,复习其临床和病理资料;所有病例均重新阅片并确诊。
1.2方法标本均经10%中性福尔马林固定,常规脱水,石蜡包埋,4 μm厚切片,行HE、免疫组化EnVision法染色及EBER原位杂交检测。CD2、CD5、CD3、CD4、CD7、CD8、CD20、CD30、CD43、CD56、Pax-5、TIA-1、Perforin、GranzymeB、Ki-67,抗体及试剂盒均购自广州安必平公司。EBER试剂盒购自北京中杉金桥公司。TCR基因重排均由外院完成。
2 结果
2.1临床特点6例患者均为儿童,其中男童4例,女童2例,初始症状出现年龄为2~11岁,病史1个月~4年。临床表现为无明显诱因出现四肢、颜面部等曝光部位水痘样疱疹或红色斑丘疹,部分有溃疡,可自行消退,遗留瘢痕,但反复发作,后逐渐增多,蔓延至大腿、上肢、躯干部等非曝光部位。6例患儿均出现反复发热,最高体温41 ℃,予消炎退热治疗后反复。辅助检查示EBV衣壳抗原VCA-IgA抗体及早期抗原EA-IgA抗体阳性,EBV-DNA拷贝数明显升高,多为105~106copy/mL(参考值为<103copy/mL)。3例患儿行PET/CT检查示皮肤多发病灶,皮肤增厚,代谢活跃,并全身多个淋巴结代谢较活跃考虑浸润。3例患儿未查见肝脾肿大等,其余3例情况不明。其中5例于2016年前确诊为HVLL,3例患儿行化疗(吉西他滨、依托泊苷+长春新碱等6~7个疗程)。1例患儿使用甲强龙+阿奇霉素+环孢素治疗,治疗后均发现皮疹较前缩小、减少,但治疗间期又有少量新发病灶出现。1例患儿治疗情况不详。另1例于2016年确诊为HV样淋巴组织增生性疾病,予小剂量甲泼尼松龙片、阿昔洛韦片口服。随访6例患儿其中5例获得随访资料,1例失访。3例2016年前诊断的患儿均已停止用药1年以上,患儿一般情况可,2例诉仍有少数新发水疱出现,可自行消退,无发热;1例诉无水疱出现(甲强龙+阿奇霉素+环孢素治疗)。1例2016年诊断的患儿仍门诊随访。1例患儿死亡。
2.2镜检送检标本均为小块皮肤组织,1例可见表皮局灶性松解,水疱形成(图1),2例可见局灶表皮溃烂,多量中性粒细胞及浆液渗出,组织坏死累及真皮浅层。表皮内未见淋巴细胞浸润,真皮层至皮下脂肪内见数量不等的淋巴细胞、中性粒细胞、嗜酸性粒细胞、组织细胞浸润,多位于小血管及附属器周围。淋巴细胞胞体中等偏小,轻至中度异型(图2),胞质淡染,核膜不规则,核分裂偶见,未见明显血管侵犯。个别小血管纤维素性坏死。
2.3免疫表型大多数淋巴细胞表达CD2、CD3、CD5、CD7、CD43、TIA-1,部分病例表达Perforin、Granzyme B。1例CD4、CD8阳性细胞数量相当,3例CD4阳性细胞占显著优势,CD8仅为少数散在阳性细胞;1例仅表达CD4,不表达CD8;1例CD8少数细胞阳性,CD4阴性。仅1例CD56阳性,1例CD30阳性。所有病例均不表达CD20、Pax-5。Ki-67增殖指数为10%~70%,中位数为42.3%。
2.4EBER与TCR检测6例均行EBV原位杂交检测,其中5例为阳性,1例为阴性。阳性细胞数目不等,且差异较大(图3)。3例患儿加做TCR分子检测,其中2例T细胞淋巴瘤克隆性基因重排阳性(图4),1例因DNA浓度太低无法检测。

①②③图1 镜下见表皮内水疱形成图2 浸润的淋巴样细胞轻度异型图3 部分病例EBER阳性细胞近100%,原位杂交法
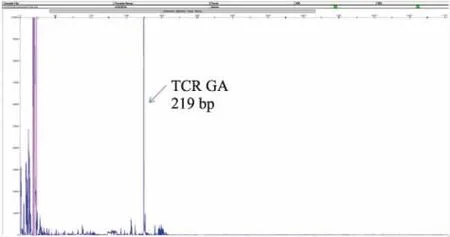
图4 TCR检测见TCR GA扩增,片段长为219 bp
3 讨论
EBV是人疱疹病毒4型,为双链DNA病毒。人体初次感染EBV通常是无症状,多发于儿童时期。在正常的宿主体内,初次感染后EBV通常潜伏在B淋巴细胞内。少数EBV可感染先天性免疫缺陷患者的T淋巴细胞或NK细胞,可以导致特发性的儿童淋巴组织增生性疾病。儿童原发性感染可以导致嗜血细胞综合征或暴发性系统性T淋巴细胞增殖性疾病。部分患者可发展为慢性活动性EBV(chronic active EBV, CAEBV)感染型T/NK细胞增殖性疾病,包括系统性CAEBV感染、HV样淋巴组织增生性疾病和蚊虫叮咬过敏[3]。
在CAEBV感染的情况下,EBV潜伏于宿主体内,可持续复制增生,导致一系列临床症状,包括长期间断发热,肝、脾、淋巴结肿大,皮肤损害等,外周血病毒DNA载量升高,EBV抗体滴度升高。CAEBV感染患者可表现为T淋巴细胞、NK细胞或B淋巴细胞的单克隆、寡克隆和多克隆增生,其中以T淋巴细胞和NK细胞的单克隆增生最多见[4-5]。有学者观察CAEBV感染患者,根据增生淋巴样细胞的免疫表型及是否存在单克隆增生,将患者分为4种类型[6],提示该类疾病的复杂原理及其必然导致的复杂临床表现。
WHO(2008)淋巴瘤分类中,将HVLL确认为独立的淋巴瘤类型。近年的临床实践中,众多病理、临床医师逐渐认识到如同CAEBV感染的复杂细胞增生状态、免疫表型,该类疾病应为独立疾病谱系[7-8],细胞可为单克隆增生,也可为多克隆增生,疾病的生物学进程可为良性、交界性和恶性,甚至可以相互进展[9]。虽然有患者转变为系统性淋巴瘤,但许多患者长期处于淋巴细胞增生状态,不能简单地全部视为淋巴瘤。WHO(2016)淋巴组织肿瘤分类中接受了该观点:考虑到该类疾病与CAEBV的关系,以及其临床过程可能为独立的疾病谱系,将HVLL改名为HV样淋巴组织增生性疾病,包括具T细胞和NK细胞表型的HV、非典型HV及HVLL。
HV属于少见的慢性特发性光敏感性皮肤病,主要特征是日晒后于躯体曝光部位出现红斑、水疱,中央有脐窝,可能伴疼痛、瘙痒,3~4天后糜烂、结痂,愈合后遗留点状凹陷性瘢痕和色素沉着。徐子刚等[10]报道12例中国HV患儿,证实皮损中存在数量不等的被EBV感染的细胞,提示其发病机制可能与EBV感染有关。HV多于儿童期发病,春夏加重,冬季减轻,病程迁延数年,约2/3患者在青春期后逐渐痊愈。临床表现轻,皮损只限于暴露部位,组织病理改变主要局限于表皮层和真皮浅层,多不伴系统症状。HV样淋巴组织增生性疾病的初始临床症状与HV十分相似,但患儿的皮损与日光照射无关,皮损更大而深在,可累及真皮深部至皮下脂肪组织,可引起大量的组织缺失而致变形;且患儿多伴发热等系统症状。组织学上表现为异型程度不等、数量不等的淋巴样细胞浸润于真皮层至皮下组织层,多位于血管及附属器周围,并可形成脂膜炎样改变。本组病例未见血管破坏及血管炎。有学者报道病灶可伴有多量嗜酸性粒细胞浸润[8],并可见血管侵犯[11]。免疫组化证实浸润的细胞可为T细胞或NK细胞,以T细胞更为多见,本组仅1例CD56呈阳性。多可表达多种T细胞抗原及细胞毒性分子如TIA-1等,CD20等B淋巴细胞抗原均为阴性。Ki-67增殖指数差异较大,本组为10%~70%。EBER大多数病例为阳性,提示与EBV感染有关,但阳性细胞比例差异较大。部分病例TCR检测见克隆性重排。在临床医检工作中,类似病例诊断需密切结合临床,了解患儿的病史及相关实验室检查结果,包括EBV抗体水平及EBV-DNA拷贝数等。需警惕的是,浸润的淋巴样细胞可能数量较少,异型性可能较轻,行免疫组化Ki-67增殖指数可能较低,但结合病史仍需考虑该疾病可能。可以加做EBER原位杂交检测了解病变部位是否存在EBV感染,TCR分子检测是否出现TCR克隆性重排可帮助诊断。
由于该病镜下形态差异较大,需综合考虑以下几点联合判断:(1)细胞的异型性愈加明显,核分裂增多,浸润范围扩大;(2)原可表达多种T细胞抗原的肿瘤细胞转变为仅表达一种或少数几种;(3)Ki-67增殖指数明显升高;(4)TCR克隆性重排阳性。同时结合临床患儿是否出现新发水疱,病变范围是否扩大,是否继续原治疗期间症状无好转甚至加重,外周血EBV抗体滴度及EBV DNA拷贝数是否持续增高,是否出现其他系统症状等,有助于鉴别。本组6例患者中2例淋巴样细胞密集丰富,较异型,弥漫浸润至皮下脂肪内,免疫组化标记1例仅表达CD4,不表达CD8;1例CD8少数细胞阳性,CD4阴性;Ki-67增殖指数分别为10%、40%,TCR克隆性重排阳性,笔者认为应归为恶性;3例患者淋巴样细胞为轻度或轻至中度异型,数量较少或中等,多位于真皮小血管及附属器周围,亦见于皮下脂肪浅部,CD4阳性细胞占显著优势,CD8仅为少数散在阳性细胞,Ki-67增殖指数10%~70%,未行TCR检测,患儿病程1~6年,均有治疗后缓解但反复发作病史,笔者认为其属于交界性病例;1例患者病史仅为1个月,浸润细胞数量少,轻度异型,CD4、CD8阳性细胞数量一致,Ki-67增殖指数约20%,EBER阳性细胞数目仅为少数散在,笔者认为性质未明,仍需临床观察了解转归判断性质。
此外,HV样淋巴组织增生性疾病还需与其他皮肤淋巴瘤鉴别:(1)发生于皮肤的结外NK/T细胞淋巴瘤,该病也可伴EBV感染,免疫组化也可出现T细胞抗原及CD56、细胞毒性分子阳性。但该病变多发于躯干和四肢,临床表现为皮肤肿块,侵袭性明显,病情进展迅速,镜下见皮肤全层受累,瘤细胞多为中等大小,异型性明显,坏死及血管浸润多见,并可浸润表皮及腺体上皮形成Pautrier微脓肿样改变,免疫组化标记CD56弥漫强阳性,CD5多为阴性,多数病例为TCR Ig基因重排阴性。(2)皮下脂膜炎样T细胞淋巴瘤,临床多表现为多发性皮下结节,无水疱。镜下见皮下脂肪中可见瘤细胞弥漫性浸润,形成小叶性脂膜炎样改变。真皮和表皮一般不受侵犯。瘤细胞呈多形性,有价值的诊断依据为瘤细胞围绕在单个的脂肪细胞周围呈环形排列。免疫组化标记CD4多为阴性,CD8阳性,需注意该病变EBER在西方为阴性,国内部分病例为阳性。(3)蕈样肉芽肿,病程多较长,多发于四肢,临床表现为红斑结节,镜下表现为特征性的亲表皮性浸润及表皮内Pautrier微脓肿形成,瘤细胞缺乏EBV感染,表达CD4,多不表达CD8及细胞毒性分子等可以区别。(4)皮肤间变大细胞淋巴瘤,病程较长,进展缓慢,多发于四肢,表现为单发或多发性皮肤结节。瘤细胞为间变性细胞,有明显的嗜酸性核仁。瘤细胞表达CD30及细胞毒性分子,缺乏EBV感染等可区别。
由于HV样淋巴组织增生性疾病临床过程多样,不同患者可能处于不同的淋巴组织增生状态,并无标准治疗方案,多强调个体化治疗。本组患者接受吉西他滨化疗和接受糖皮质激素治疗的患儿均取得临床缓解。目前,文献报道[12-13]的治疗方案有放、化疗、大剂量母体淋巴细胞输注、中药、免疫调节及抑制剂等,多可取得短期缓解。
HV样淋巴组织增生性疾病的预后,各报道差异较大。由于本组病例数较少,我们检索2009年至今发表的30余篇相关文章,均为个案报道(55例),其中儿童49例,成人9例,该病好发于女童;死亡7例,病死率为12.7%,低于本组数据20%,但病例数仍较少,还需更多资料来帮助判断其预后。可以预测的是,停留在淋巴组织增生状态的患儿,预后较好。但对于出现淋巴细胞单克隆增生,向系统性淋巴瘤转化的患儿,预后较差。目前,认为EBV感染活跃程度,发病年龄大于8岁、合并血液系统症状、T细胞来源及系统损害是影响预后的主要危险因素[14]。
综上所述,HV样淋巴组织增生性疾病较为少见,与慢性活动性EBV感染密切相关,其临床过程可能为独立疾病谱系,其性质可为良性、交界性及恶性。临床需密切随访观察,病理诊断需密切结合临床、HE形态、免疫组化及分子检测。该病治疗及预后仍需积累更多资料,探索更加积极有效的治疗及预防手段。
[1] Sabattini E, Bacci F, Sagramoso C,etal. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview[J]. Pathologica, 2010,102(3):83-87.
[2] Swerdlow S H, Campo E, Pileri S A,etal. The 2016 revison of the world health organization clsssificaton of lymphoid neoplasm[J]. Blood, 2016,127(20):2375-2390.
[3] Sanghui P, Young H K. Epstein-Barr virus-associated T/natural killercell lyphoproliferative disorders[J]. J Deratol, 2014,41(1):29-39.
[4] Cohen J I, Kimura H, Nakamura S,etal. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008[J]. Ann Oncol, 2009,20(9):1472-1482.
[5] Suzuki K, Ohshima K, Karube K,etal. Clinicopathological states of Epstein-Barr virus-associated T/NK-cell lymphoproliferative disorders (severe chronic active EBV infection of children and young adults[J]. Int J Oncol, 2004,24(5):1165-1174.
[6] Ohshima K, Kimura H, Yoshino T,etal. Proposed categorization of pathological states of EBV-associated T/natural killer-cell lymphoproliferative disorder (LPD) in children and young adults: overlap with chronic active EBV infection and infantile fulminant EBV-T-LPD[J]. Pathol Int, 2008,58(4):209-217.
[7] 王 觅,高立敏,杨群培. 种痘水疱病样T细胞淋巴瘤的临床病理特征分析[J]. 西部医学, 2016,28(11):1558-1565.
[8] 罗 颖,黄海燕,杜 鹃,等. 伴有嗜酸性粒细胞增多的种痘样水疱病样EB病毒感染相关淋巴细胞增生性疾病[J]. 临床皮肤科杂志, 2014,43(11):659-662.
[9] 周小鸽,何乐建,金 妍. EB病毒淋巴增殖性疾病国际分类会议介绍及分类说明[C]. 天津:第十一届中国抗癌协会全国淋巴瘤学术大会教育论文集, 2009:107-118.
[10] 徐子刚,马 琳,申昆玲,等. 牛痘样水疱病与Epstein-Barr病毒潜伏感染的关系[J]. 中华皮肤科杂志, 2005,38(4):238-239.
[11] 曹雪萍,徐教生,徐子刚. 种痘样水疱样EB病毒感染相关淋巴细胞增生性疾病一例[J]. 实用皮肤病学杂志, 2015,8(5):394-396.
[12] Beltrán B E, Maza I, Moisés-Alfaro C B,etal. Thalidomide for the treatment of hydroa vacciniforme-like lymphoma: report of four pediatric cases from Peru[J]. Am J Hematol, 2014,89(12):1160-1161.
[13] 周 令,徐学聚,张 园,等. 儿童种痘水疱病样淋巴瘤二例报告附文献复习[J]. 中华血液学杂志, 2013,34(6):485-488.
[14] 张秋鹂,汪 旸,涂 平. 成人种痘样水疱病样EB病毒相关淋巴细胞增生性疾病三例[J]. 中国麻风皮肤病杂志, 2016,32(4):203-206.
Clinicalandpathologicalanalysisonhydroavacciniforme-likelymphoproliferativedisorderinchildren
FU Si-ying1, QI Qing2, LI Qiu-yan2, KUANG Zhong-sheng1, XIE Yu-hui1
(1DepartmentofPathology,2DepartmentofDermatology,theFirstAffiliatedHospitalofGuangzhouUniversityofTCM,Guangzhou510405,China)
PurposeTo discuss the clinical, histopathological characteristics, diagnosis, differential diagnosis and prognosis of hydroa vacciniforme-like lymphoproliferative disorder in children.Methods6 cases of hydroa vacciniforme-like lymphoproliferative disorder were analyzed by molecular, histopathological and immunohistochemical testing. Clinical and follow-up information was obtained. The published relevant literatures were reviewed.Results4 cases were boys, 2 case were girls. All the patients presented with erythema and blisters with fever for 1 month to 4 years. Histopathologic examination showed an mild or moderate atypical lymphocytic infiltrate with angiotropism and angiocentricity, and scattered or dense lymphoid infiltration throughout the dermis and subcutaneous tissue. Blisters or necrosis could be seen in the epidermis. The atypical lymphocytes were positive for CD2, CD3, CD5, CD7, CD43, TIA-1, CD4 or CD8, and negative for CD20, Pax-5. Only one case showed positive stain for CD56. The average positive rate of Ki-67 in tumor cells was 42.3%. Tumor cells positive for EBV encoded RNA (EBER) were detected in cutaneous infiltrates in 5 cases. Gene rearrangement of TCR was detected in 2 cases. 5 patients were available for follow-up examination and 1 patient was dead.ConclusionHydroa vacciniforme-like lymphoproliferative disorder is a rare disease mainly occuring in children. Chronic active EBV infection has been associated with this disease. It may be a spectrum in terms of its clinical course, and may be benign, borderline and malignant. Pathological diagnosis should be closely combined with clinical data.
hydroa vacciniforme; lymphoma; Epstein-Barr virus; clinicopathological feature
时间:2017-9-18 6:23 网络出版地址:http://kns.cnki.net/kcms/detail/34.1073.R.20170918.0623.014.html
R 739.5
A
1001-7399(2017)09-1005-05
10.13315/j.cnki.cjcep.2017.09.014
接受日期:2017-07-16
广州中医药大学第一附属医院1病理科、2皮肤科,广州 510405
傅思莹,女,硕士,主治医师。Tel:(020)36591877,E-mail: babeque05@163.com
