二苯并[b,f][1,5]二氮杂芳辛四烯衍生物的合成及拆分
2017-09-16李正义钱纪生朱美兰孙小强
李正义, 钱纪生, 朱美兰, 潘 勇, 殷 乐, 孙小强*
(1. 常州大学 石油化工学院,江苏 常州 213164; 2. 常州四药制药有限公司,江苏 常州 213004)
·研究论文·
二苯并[b,f][1,5]二氮杂芳辛四烯衍生物的合成及拆分
李正义1, 钱纪生1, 朱美兰2, 潘 勇1, 殷 乐1, 孙小强1*
(1. 常州大学 石油化工学院,江苏 常州 213164; 2. 常州四药制药有限公司,江苏 常州 213004)
以邻氨基二苯甲酮为原料,经自身缩合环化合成了3种二苯并[1,5]二氮杂芳辛四烯衍生物(1a~1c);以邻苯二甲酸酐和溴苯为原料经傅-克反应制得中间体2-(4-溴苯甲酰溴)苯甲酸(M1);M1经叠氮化后自缩合制得6,12-二(4-溴苯基)二苯并[b,f][1,5]二氮杂环辛四烯(1d);以邻氨基苯甲酸甲酯为原料,经自身缩合环化制得中间体二苯并[b,f][1,5] 二氮杂环辛四烯-6,12(5H,11H)-二酮(M2);M2经氯化合成6,12-二氯二苯并[b,f][1,5]二氮杂环辛四烯(1e),化合物1a~1e的结构经1H NMR,13C NMR和ESI-MS表征,其中化合物1c为新化合物。利用超临界色谱(SFC)技术对化合物1a~1e实现了手性拆分,获得5对具有高旋光度的光学活性异构体(ee>99%)。
邻氨基二苯甲酮; 邻苯二甲酸酐; 邻氨基苯甲酸甲酯; 二苯并[b,f][1,5]二氮杂芳辛四烯; 合成; 手性拆分
二苯并[b,f][1,5]二氮杂芳辛四烯及其衍生物是八元大环结构[1-2]中的一类经典代表[3],其结构中含有两个具有配位性能的碳氮双键,且其母体骨架的可修饰性,使其具有良好的配位性能[4-5],已广泛应用于催化反应[6-8]、功能材料[9-11]及电化学[12-13]等领域。二苯并[1,5]二氮杂芳辛四烯衍生物还具有抗促性腺激素、降血压、降血脂胆固醇和雌性激素等生物和药物活性[14-15],因此该类化合物的合成及应用一直是研究人员关注的热点。1896年,Sondheimer等[16]首次利用邻氨基二苯甲酮合成了二苯并[b,f][1,5]二氮芳辛四烯衍生物,但由于原料底物的局限性,此类化合物的合成受到了限制。随后,新的合成方法相继出现,例如用二苯酮类叠氮化合物经过Curtiu 重排形成烯碳进而环化脱羧形成二苯并[b,f][1,5]二氮杂环类化合物[17-19],但此法反应步骤增多,反应条件苛刻且后处理繁琐。因此,二苯并[1,5]二氮杂芳辛四烯的合成方法仍有待进一步开发。更为有趣的是该类化合物与Tröger’s碱[20]类似具有刚性的八元环骨架,因而具有稳定的构象,由此可以产生稳定的面手性,但其光学异构体的拆分和光学活性尚无人报道。
本文通过3种方法合成了5种具有不同取代基的二苯并[b,f][1,5]二氮杂芳辛四烯衍生物(Scheme 1):以取代邻氨基二苯甲酮为原料,在无溶剂条件下,经自身缩合环化制得3种二苯并[1,5]二氮杂芳辛四烯衍生物(1a~1c);以邻苯二甲酸酐和溴苯为原料经傅-克反应制得2-(4-溴苯甲酰溴)苯甲酸(M1);M1经叠氮化后自缩合得6,12-二(4-溴苯基)二苯并[b,f][1,5]二氮杂环辛四烯(1d);以邻氨基苯甲酸甲酯为原料自身缩合环化制得二苯并[b,f][1,5]二氮杂环辛四烯-6,12(5H,11H)-二酮(M2);M2经氯化合成6,12-二氯二苯并[b,f][1,5]二氮杂环辛四烯(1e),化合物1a~1e的结构经1H NMR,13C NMR和MS(ESI)表征。运用超临界色谱技术成功对其进行了拆分,获得了5对光学纯度均大于99%ee的光学纯异构体。
1 实验部分
1.1 仪器与试剂
SGW X-4型显微熔点仪(温度未校正);VANCE 300 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);LCMS-2020型质谱仪; Novasep-3050型超临界高压液相制备色谱仪;Chiralcel IA (SFC)硅胶表面键合直链淀粉-三(3,5-二甲苯基氨基甲酸酯)超临界流体色谱用手性制备色谱柱(Daicel, 250 mm×50 mm, 5 μm)。
所用试剂均为分析纯。
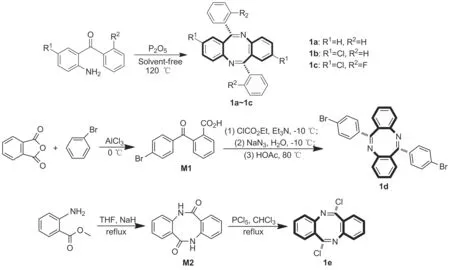
Scheme 1
1.2 合成
(1)1a~1c的合成通法[21]
在反应瓶中加入取代2-氨基二苯甲酮40 mmol与五氧化二磷8 mmol,搅拌下于120 ℃反应5 h(TLC检测反应完全)。加水20 mL,用二氯甲烷(3×30 mL)萃取,合并萃取液,依次用饱和NaCl溶液洗涤,无水Na2SO4干燥,减压除去溶剂,残余物用乙醇重结晶得1a~1c。
6,12-二苯基二苯并[b,f][1,5]二氮杂环辛四烯(1a): 淡黄色固体6.7 g,收率85%, m.p. 185~187 ℃(191~193 ℃[22]);1H NMRδ: 7.78~7.75(m, 4H, ArH), 7.40~7.25(m, 8H, ArH), 7.05~6.95(m, 6H, ArH);13C NMRδ: 169.6, 159.9, 138.0, 131.1, 129.7, 129.4, 128.2, 127.6, 126.9, 123.4, 120.9; MS(ESI)m/z: 359{[M+H]+}。
2,8-二氯-6,12-二苯基二苯并[b,f][1,5]二氮杂环辛四烯(1b): 黄色固体1.45 g,收率85%, m.p. 217~219 ℃(200~208 ℃[23]);1H NMRδ: 7.74~7.71(m, 4H, ArH), 7.46~7.29(m, 8H, ArH), 6.99~6.95(m, 4H, ArH);13C NMRδ: 168.8, 150.2, 137.2, 131.6, 130.1, 129.4, 129.1, 128.4, 128.1, 127.2, 122.5; MS(ESI)m/z: 427{[M+H]+}。
2,8-二氯-6,12-二(2-氟苯基)二苯并[b,f][1,5]二氮杂环辛四烯(1c): 黄色固体15.3 g, 收率70%, m.p. 219~220 ℃;1H NMRδ: 7.86~7.80(m, 2H, ArH), 7.48~7.40(m, 2H, ArH), 7.32~7.28(m, 2H, ArH), 7.24~7.18(m, 2H, ArH), 7.07~6.91(m, 6H, ArH);13C NMRδ: 165.9, 163.0, 159.6, 148.3, 133.1, 131.3, 130.1, 129.6, 126.0, 124.2, 122.7, 116.9, 116.6; MS(ESI)m/z: 463{[M+H]+}。
(2) 6,12-二(4-溴苯基)二苯并[b,f][1,5]二氮杂环辛四烯(1d)的合成
在单口烧瓶中加入邻苯二甲酸酐16 mmol,无水三氯化铝32 mmol和溴苯10 mL,搅拌使其溶解;于0 ℃反应4 h。加入1 mol·L-1盐酸(产生白色固体),抽滤,滤饼用20% NaOH溶液溶解,用1 mol·L-1盐酸调节pH至4~5(析出白色固体),抽滤,滤饼干燥得中间体M1。
将中间体M1溶于THF(20 mL)中,搅拌下于-10 ℃滴加三乙胺1 mL,滴毕(30 min),再滴加氯甲酸乙酯0.67 mL,滴毕(30 min),反应2 h;加入NaN30.24 g的水(14 mL)溶液,反应6 h;自然升至室温,加入乙酸5 mL,升温至80 ℃,反应6 h。用饱和Na2CO3溶液调至中性,用二氯甲烷(3×10 mL)萃取,合并萃取液,用饱和NaCl溶液洗涤,无水Na2SO4干燥,减压除去溶剂,残余物用乙醇重结晶得黄色固体1d1.3 g, 收率72%, m.p. 235~237 ℃;1H NMRδ: 7.64~7.32(m, 10H, ArH), 7.06~6.92(m, 6H, ArH);13C NMRδ: 168.6, 151.5, 136.7, 131.5, 130.9, 129.9, 127.4, 126.2, 126.1, 123.7, 120.9; MS(ESI)m/z: 516{[M+H]+}。
(3) 6,12-二氯二苯并[b,f][1,5]二氮杂环辛四烯(1e)的合成
将邻氨基苯甲酸甲酯26.4 mmol和氢化钠52.8 mmol溶于50 mL无水THF中,搅拌下回流反应至终点(TLC监测)。用1 mol·L-1盐酸调节pH至4~5(析出固体),抽滤,滤饼用乙醇重结晶得中间体M2。
将中间体M28 mmol和五氯化磷24 mmol溶于氯仿20 mL中,搅拌下回流反应4 h(TLC监测)。旋蒸除去溶剂,残余物经硅胶柱层析[洗脱剂:V(乙酸乙酯)/V(石油醚)=1/10]纯化得白色固体1e0.9 g,收率45%, m.p.207~209 ℃;1H NMRδ: 7.40~7.29(m, 4H, ArH), 7.19~7.08(m, 2H, ArH), 7.00~6.91(m, 2H, ArH);13C NMRδ: 156.3, 145.3, 131.6, 127.1, 126.1, 125.4, 122.0; MS(ESI)m/z: 297{[M+Na]+}。
1.3 二苯并[1,5]二氮杂芳辛四烯衍生物的手性拆分
分别称取适量1a~1e,用流动相[V(液态CO2)/V(异丙醇/二氯甲烷)=85/15(80/20)]溶解得0.002 g·mL-1样品溶液,设置流动相流速为2.0 mL·min-1,检测波长为220 nm,柱温箱温度为35 ℃;取上述样品溶液2 mL注入超临界高压液相制备色谱仪,收集两种异构体馏分,重复上述步骤,循环操作100 次,将收集到的两种异构体的所有馏分低温蒸除溶剂,真空干燥,分别得到两种光学异构体各100 mg。
2 结果与讨论
2.1 合成
二苯并[b,f][1,5]二氮杂芳辛四烯衍生物中与八元环骨架相连或相并苯环上取代基团对该类化合物的合成途径选择具有重要的影响,本文采用3种不同方法合成了5种具有不同取代基团的二苯并[b,f][1,5]二氮杂芳辛四烯衍生物。
采用传统合成方法,如Scheme 1所示,以邻氨基二苯甲酮类化合物为原料,在无溶剂条件、P2O5作用下发生分子间的脱水缩合环化反应,制得化合物1a~1c。此方法简便、易操作、副产物少且收率较高(70%~85%),但各种取代邻氨基二苯甲酮不易获得,且氨基的活性受基团影响较大,因此该方法的底物局限性较大。
以邻苯二甲酸酐和溴苯为原料,在三氯化铝作用下制得2-(4-溴苯甲酰溴)苯甲酸(M1),中间体(M1)在氯甲酸乙酯、三乙胺和叠氮化钠作用下将羧基进行叠氮化后,在乙酸中完成双分子缩合合成1d,两步反应总收率72%。
另外,以邻氨基苯甲酸甲酯为原料,在NaH作用下通过氨酯交换反应发生自身缩合环化得到中间体M2;M2在氯仿中经五氯化磷卤化合成1e,两步反应总收率为45%。
2.2 二苯并[b,f][1,5]二氮杂环辛四烯衍生物的拆分
由于化合物1a~1e在一般有机溶剂中的溶解性较小,无法用常规的高效液相色谱法进行拆分。本文选用掺杂有机溶剂的液态二氧化碳[V(液态CO2)/V(异丙醇/二氯甲烷)=85/15(80/20)]作为展开剂,运用超临界色谱技术成功对其进行了拆分,获得了5对光学纯度均大于99%ee的光学纯异构体。
化合物1a~1e的消旋体及对应光学异构体的比旋光度和熔点数据见表1所示。首先,该类化合物消旋体的熔点均高于其对应的两种光学异构体的熔点。其次,该类化合物的比旋光度较高,尤其是化合物1a的两种光学异构体的比旋光度分别达到了+1 923°和-1 916°。但是当与八元环骨架相连或相并的苯环上有取代基时(1b~1d),两种光学异构体的比旋光度值降低,尤其是当八元环骨架上没有取代苯基时(1e),比旋光度值急剧下降,仅为+251°和-247°。由此可见,取代基的类型对二苯并[b,f][1,5]二氮杂环辛四烯衍生物的旋光性能具有重要的影响。
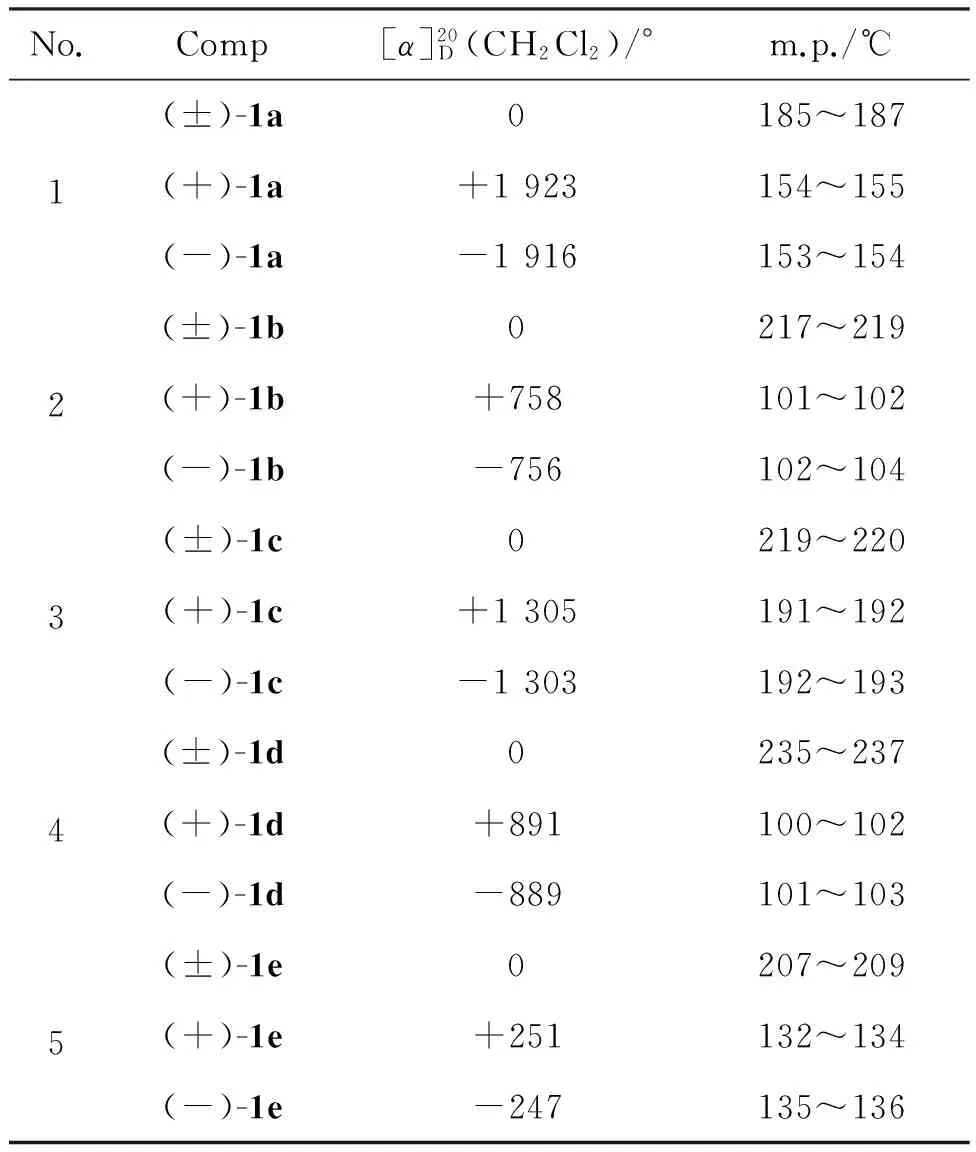
表1 化合物1a~1e的比旋光度和熔点
分别以邻氨基二苯甲酮、邻苯二甲酸酐和溴苯、邻氨基苯甲酸甲酯为原料,采用3种方法合成了5种二苯并[1,5]二氮杂芳辛四烯衍生物。运用超临界色谱技术成功对其进行了拆分,得到了5对光学纯度均大于99%ee的光学纯异构体。比旋光度数据表明,该类化合物具有较好的旋光性能,但取代基的类型对其旋光性能具有重要的影响。该研究为进一步拓展其在手性领域的应用提供了重要参考依据和数据。
[1] Heinz W, Räder H J, Müllen K. Changing the size of a cavityviaan electron-transfer:Synthesis and reduction of 1,5,22,26-tetraoxa-[5,5]-(2,8)-dibenzo[a,e]cylooctatetraenophane[J].Tetrahedron Lett,1989,30(2):159-162.
[2] Marsella M J, Reid R J, Estassi S,etal. Tetra[2,3-thienylene]:A building block for single-molecule electromechanical actuators[J].J Am Chem Soc,2002,124(42):12507-12510.
[3] Pettersson B, Bergman J, Svensson P H. Synthetic studies towards 1,5-benzodiazocines [J].Tetrahedron,2013,69(12):2647-2654.
[4] Eisch J J, Yu K, Rheingold A L. 6,12-Diphenyldibenzo[b,f][1,5]diazocine as an electron-capture agent:Efficient mechanistic probe for SET processes and reagent for the oxidative dimerization of benzylic organometallics[J].Eur J Org Chem,2012,2012(16):3165-3171.
[5] Hampton C S, Harmata M. Synthesis of Tröger’s base derived ligands:PHZ-derivatized ligand with pendant donor arms[J].Tetrahedron,2016,72(40):6064-6077.
[6] Poli E, Merino E, Díaz U,etal. Different routes for preparing mesoporous organosilicas containing the Tröger’s base and their textural and catalytic implications[J].J Phys Chem C,2011,15:7573-7585.
[7] Herrmann W A, Kühn F E, Mattner M R,etal. Multiple bonds between transition metals and main-group elements,163 nitrogen-donor adducts of organorhenium(VII) oxides:Structural and catalytic aspects 1[J].J Organomet Chem,1997,538:203-209.
[8] Didier D, Sergeyev S. Thiourea derivatives of Tröger’s base:Synthesis,enantioseparation and evaluation in organocatalysis of michael addition to nitroolefins[J].Arkivoc,2009,(14):124-134.
[9] Stehlik S, Izak T, Kromka A,etal. Sensitivity of diamond-capped impedance transducer to tröger’s base derivative[J].ACS Appl Mater Interfaces,2012,4:3860-3865.
[10] Neogi I, Jhulki S, Ghosh A,etal. Bifunctional organic materials for OLEDs based on Tröger’s base: Subtle structural changes and significant differences in electroluminescence[J].Org Electron,2014,15(12):3766-3772.
[11] Neogi I, Jhulki S, Ghosh A,etal. Amorphous host materials based on tröger’s base scaffold for application in phosphorescent organic light-emitting diodes[J].ACS Appl Mater Interfaces,2015,7:3298-3305.
[12] Suga T, Wi S, Long T E. Synthesis of diazocine-containing poly(arylene ether sulfone)s for tailored mechanical and electrochemical performance[J].Macromolecules,2009,42(5):1526-1532.
[13] Shishkanova T V, Havlík M, Král V,etal. Amino-substituted Tröger’s base:Electrochemical polymerization and characterization of the polymer film[J].Electrochim Acta,2017,224:439-445.
[14] Wakankar D M, Hosangadi B D. Studies in large ring compounds:Part VI. Synthesis of 5,11-disubstituted dibenzo[b,f][1,5]diazocine-6,12(5H,11H)-diones[J].Indian J Chem 1984,23:136-139.
[15] Manda B R, Alla M, Ganji R J,etal. Discovery of Tröger's base analogues as selective inhibitors against human breast cancer cell line:Design,synthesis and cytotoxic evaluation[J].Eur J Med Chem,2014,86(86c):39-47.
[16] Sondheimer A.A compound containing a ring of eight atoms[J].Chem Ber,1896,29:1272-1275.
[17] Wang X, Li J, Zhao N,etal. A rapid and efficient access to diaryldibenzo[b,f][1,5]diazocines[J]. Org Lett,2011,13(4):709-711.
[18] Zhao N, Qiu L, Wang X,etal. Trifluoroacetic acid catalyzed dibenzodiazocine synthesis: Optimization and mechanism study[J].Tetrahedron,2012,68:9665-9671.
[19] Acharya B P, Rao Y R. Synthesis of unsymmetrical substituted 6,12-diaryldibenzo[b,f][1,5]diazocines And their precursor schiff bases[J].Synthesis,1986,(04):324-326.
[20] Kazem-Rostami M.Design and synthesis of Λ-shaped photoswitchable compounds employing Tröger’s base Scaffold[J].Synthesis,2017,49(06):1214-1222.
[21] Jung D I, Song J H, Lee E J,etal. Simple synthesis of quinolines and dibenzo[b,f][1,5]diazocines under microwave irradiation[J].Tetrahedron Lett,2009,50(42):5805-5807.
[22] Metlesics W, Resnick T, Silverman G,etal. 6,12-Diphenyldibenzo[b,f][1,5]diazocines[J].J Med Chem,1966,9(4):633-634.
[23] Metlesics W, Tavares R, Sternbach L H. The reduction products of a dibenzo[b,f][1,5]diazocine[J].J Org Chem,1966,31(10):3356-3362.
Synthesis and Resolution of Dibenzo[b,f][1,5]diazocine Derivatives
LI Zheng-yi1, QIAN Ji-sheng1, ZHU Mei-lan2, PAN Yong1, YIN Yue1, SUN Xiao-qiang1*
(1. School of Petrochemical Engineering, Changzhou University, Changzhou 213164, China; 2. Changzhou Siyao Pharmaceuticals Co., Ltd., Changzhou 213004, China)
Three dibenzo[b,f][1,5]diazocine derivatives(1a~1c) were synthesized by self-condensation and cyclization using 2-aminobenzophenone as raw material. 2-(4-Bromobenzoyl)benzoic acid(M1) was synthesized by Friedel-Crafts reaction ofo-phthalic anhydride with bromobenzene, and then 6,12-bis(4-bromophenyl)dibenzo[b,f][1,5]diazocine(1d) was formed by self-condensation ofM1. Dibenzo[b,f][1,5]diazocine-6,12(5H,11H)-dione(M2) was obtained by self-condensation and cyclization of methyl anthranilate, and then 6,12-dichlorodibenzo[b,f][1,5]diazocine(1e) was synthesized by chlorination of intermediateM2. The structures of1a~1ewere characterized by1H NMR,13C NMR and ESI-MS. Compound1cis a new compound. All the racemates of1a~1ewere successfully chiral resolved by supercritical fluid chromatography(SFC) to give enantiomers with high optical rotation, withee>99%.
2-aminobenzophenone;o-phthalic anhydride; methyl anthranilate; dibenzo[b,f][1,5]diazocine; synthesis; chiral resolution
2017-01-27;
: 2017-07-13
国家自然科学基金资助项目(21572026); 江苏省高校自然科学研究重大项目(14KJA150002); 江苏省先进催化与绿色制造协同创新中心资助项目(ACGM2016-06-05)
李正义(1979-),男,汉族,江苏扬州人,博士,主要从事有机合成及催化的研究。 E-mail: zyli@cczu.edu.cn
孙小强,教授, E-mail: sunxiaoqiang@yahoo.com
O621.3; O652.63
: ADOI: 10.15952/j.cnki.cjsc.1005-1511.2017.09.17018
