钙钛矿APbI3结构稳定性及光电性质的理论研究∗
2017-08-01刘娜危阳马新国祝林徐国旺楚亮黄楚云
刘娜 危阳 马新国 祝林 徐国旺 楚亮 黄楚云
1)(湖北工业大学理学院,武汉 430068)
2)(湖北工业大学,太阳能高效利用湖北省协同创新中心,武汉 430068)
3)(南京邮电大学理学院,南京 210046)
钙钛矿APbI3结构稳定性及光电性质的理论研究∗
刘娜1)危阳1)马新国1)2)†祝林1)徐国旺1)2)楚亮3)黄楚云1)2)‡
1)(湖北工业大学理学院,武汉 430068)
2)(湖北工业大学,太阳能高效利用湖北省协同创新中心,武汉 430068)
3)(南京邮电大学理学院,南京 210046)
(2016年7月29日收到;2016年12月4日收到修改稿)
采用基于色散修正的平面波超软赝势方法研究了钙钛矿材料APbI3结构中四种阳离子Cs+,NH+4,MA+,FA+分别处于A位时,其结构的稳定性、电子结构及光学性质.研究结果显示受阳离子种类和尺寸的影响,PbI基体骨架发生不同程度的扭曲,A位置阳离子(除Cs+外)半径越大,其与PbI基体骨架之间的作用力越强,在MAPbI3和FAPbI3中PbI6八面体显示出较大的电偶极矩.计算得出的能带结构表明,四种体系在费米能级附近的能带相似,即价带顶均由I 5p轨道组成,导带底由Pb 6p轨道和部分I 5p轨道杂化而成.电子结构和光学性质的差异主要源于PbI6八面体结构的扭曲.在四种结构体系中,CsPbI3显示出最窄的直接带隙、最小载流子有效质量和较强的光吸收能力.这些结果可为进一步深入研究钙钛矿材料在太阳能电池领域的应用提供理论指导.
钙钛矿结构材料,结构稳定性,电子结构,第一性原理
1 引 言
钙钛矿太阳能电池是基于染料敏化太阳能电池发展起来的一种新型太阳能电池,具有光电转换效率较高、制备简单和易于规模化生产等优点,掀起了这类太阳能电池的研究热潮[1−3].2009年,Kojima等[4]在染料敏化太阳能电池中首次将有机金属卤化物CH3NH3PbX3(X为Br,I)作为光吸收剂,制作的太阳能电池器件获得了3.8%的光电转换效率.之后,有机金属卤化物作为钙钛矿太阳能电池的关键材料引起了人们极大的关注.尽管在提高钙钛矿太阳能电池器件光电转换效率方面的研究进展十分迅速[5],但是要实现钙钛矿太阳能电池的商业化,除了提高光电转换效率外,更大的挑战在于解决电池的稳定性问题[6−8],尤其是作为光吸收层的钙钛矿材料的稳定性.
钙钛矿材料表现出较强的光吸收能力和较高的载流子迁移率,源于其特殊的有机无机杂化结构,分子式为ABX3,其中BX6构成八面体,并相互接触组成了三维结构框架,带电的A离子嵌入其中[9].其稳定性能通过容差因子t进行判断,其中RA,RB和RX分别表示A,B和X原子的有效半径.目前对钙钛矿材料CH3NH3PbX3体系的研究较多,为了提高其结构稳定性和光电转换性能,基于该结构中A位置的阳离子替代的改性研究已经展开[10−14].如选取离子半径更大的替代形成的结构晶格常数更大,带隙稍小[10−12].尽管利用该钙钛矿材料制作的太阳能电池的效率达到20%以上,但是其结构稳定性较差,严重影响了实际应用.最近,部分实验采用Cs+替代A位置MA+,发现其量子效率并没有降低,同时钙钛矿材料的结构稳定性更高,显示出极大的应用潜力[13,14].
目前大量的研究主要集中在钙钛矿材料的合成和掺杂改性等工艺上[15],但是因其结构的特殊性和A位阳离子与PbI6骨架之间较弱的相互作用,很难通过实验直接探测,其微结构与电子结构之间的关联性仍然需要进一步深入研究[16].采用第一性原理的方法可以获得实验研究无法获取的有用信息,从材料设计的角度分析微结构对光电性能的影响.最近有研究人员采用该方法研究了这类材料的电学性质[17−21],但是有机阳离子的种类对结构稳定性和光电性质影响的系统研究仍然没有展开,其较强的光吸收能力和较高的载流子迁移率的物理机理仍然不清楚.为此,我们在A位选取不同的阳离子建立钙钛矿物理模型,即A位为Cs+,NH+4,MA+和FA+,采用基于密度泛函理论的第一性原理方法深入研究阳离子对钙钛矿结构稳定性、电子结构和光学性质的影响,为进一步提高钙钛矿材料的光吸收能力和光电转换效率提供理论指导.
2 物理模型与计算方法
选取MAPbI3室温下的四方相(空间群为I4/mcm)为研究对象,一个晶胞由4个MAPbI3分子组成,共48个原子,初始晶格常数采用实验数据a=b=0.880 nm,c=1.269 nm[22].在此结构的基础上,分别用取代MAPbI3结构中A位置的有机阳离子MA+,获得了CsPbI3,NH4PbI3和FAPbI3的初始晶体结构,再分别对其进行几何结构优化.A位置的四种阳离子分别为和FA+,其分子构型如图1所示.在MAPbI3和FAPbI3结构中,考虑了MA+和FA+的不同取向对体系总能量的影响,而电子结构计算仅研究了对应结构中体系总能量最低的结构取向情形.
采用基于密度泛函理论的平面波超软赝势方法[23],考虑阳离子基团和PbI骨架之间的相互作用,分别在广义梯度近似(GGA)的PBE方案中考虑了TS色散修正[24],在局域密度近似(LDA)的CAPZ方案中考虑了OBS色散修正[25],获得了相应的晶格常数、Pb—I键长、能带结构及光学性质等.在描述离子实与价电子之间的相互作用时,选取的价电子组态分别为C:2s22p2,H:1s1,N:2s22p3,Pb:5d106s26p2,I:5s25p5,Cs:5s25p66s1.布里渊区k点网格均选为4×4×3[26]. 采用BFGS算法对所有模型进行几何结构优化,平面波截断能设置为400 eV,自洽收敛精度设置为5.0×10−5eV/atom,原子间的力场收敛精度设置为0.01 eV/nm,最大应力设置为0.2 GPa,最大位移设置为5×10−4nm.所有计算均由CASTEP软件完成[27].
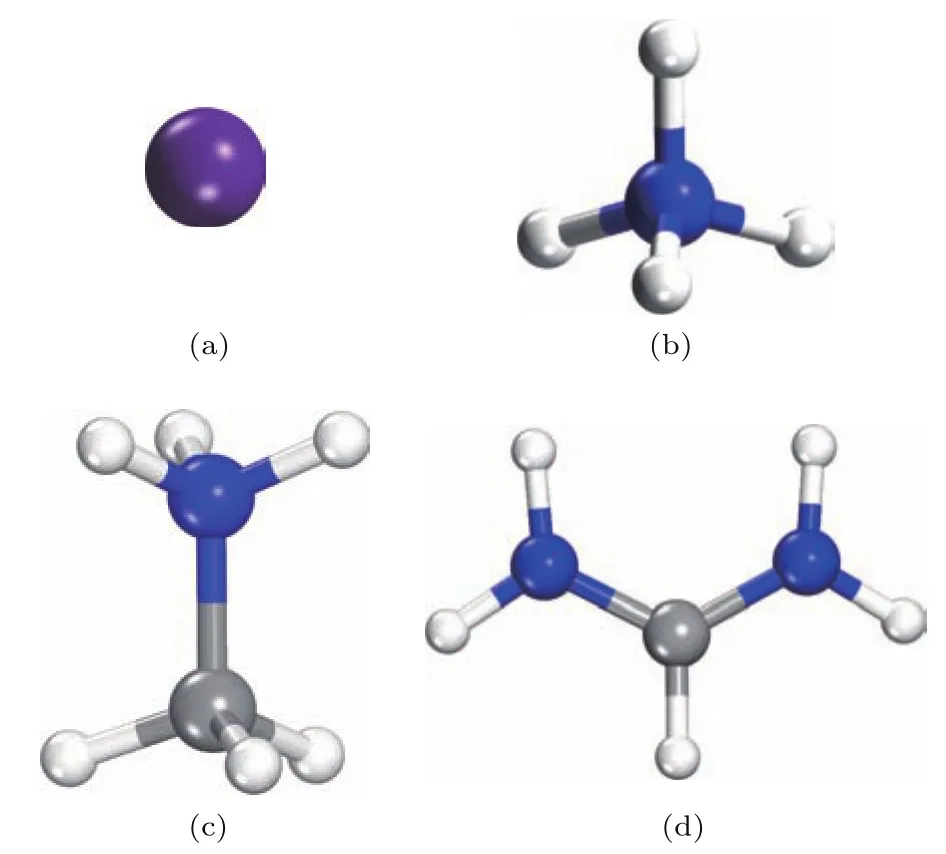
图1 (网刊彩色)钙钛矿APbI3结构中A位阳离子的分子构型(紫色球为Cs原子,蓝色球为N原子,灰色球为C原子,白色球为H原子) (a)Cs+;(b)NH;(c)CH3NH;(d)为CH(NH2)Fig.1. (color online)Four molecule models of A cations in perovskite APbI3(purple ball is Cs atom,blue balls are N atoms,gray balls are C atoms,and white balls are H atoms):(a)Cs+;(b)NH;(c)CH3NH;(d)CH(NH2).
3 结果与讨论
3.1 结构稳定性
为了获得结构合理且总能量较低的稳定晶体结构,对建立的所有结构进行全局几何结构优化,即不固定原子位置、晶格常数以及各棱之间夹角α,β,γ,优化后的晶体结构如图2所示.可以看出,结构优化后NH4PbI3,MAPbI3,FAPbI3中PbI6八面体在一定程度上发生了较明显的倾斜和旋转,而在CsPbI3中PbI6八面体的倾斜和旋转不明显.表1为采用基于TS色散修正的GGA(PBE)方法和OBS色散修正的LDA(CAPZ)方法对4种钙钛矿结构进行全局结构优化后,获得的晶体结构参数、禁带宽度和键长等结果.
由于Cs原子与周围PbI骨架之间是共价键作用,不存在较弱的范德瓦耳斯力,因此对CsPbI3体系进行几何结构优化时没有考虑色散修正.众所周知,LDA计算的晶格常数比实验值偏小,而GGA计算的晶格常数比实验值偏大.例如,在MAPbI3结构中分别采用GGA和LDA方法,计算出的晶胞体积分别比实验值大7.54%和小7.05%.由此可见计算方法的选择在一定程度上影响晶体结构和电子结构的结果.总体来说,除了Cs+外,晶胞体积会随着有机阳离子FA+半径RFA+=0.279 nm)的增大而增大.由于Pb2+和I−离子的半径分别为0.133 nm和0.203 nm,稳定的钙钛矿结构中占据A位置的粒子半径RA应该满足0.130 nm
表1列出了采用GGA(PBE)和LDA(CAPZ)两种方法获得的Pb—I键长以及H和I之间的距离,这里的“数量”为H和I间距小于0.28 nm的近邻数目.可以看出,与其他体系相比,在CsPbI3中Pb—I键长的平均值最小,且Pb—I键长的变化范围最小,这表明PbI6构成的八面体扭曲及旋转程度最小.随着A位阳离子(除Cs+)有效半径的增大,在GGA算法中,Pb—I的平均键长从0.325 nm增大至0.329 nm,H和I之间的近邻数目从7增大至11;而在LDA算法中,Pb—I的平均键长从0.316 nm增大到0.320 nm,H和I之间的近邻数目从17增大到21.这说明阳离子A的半径越大,与无机骨架之间的作用力将越强.在MAPbI3中Pb—I键长的变化范围分别为0.046 nm(GGA)和0.064 nm(LDA),其对应的PbI6八面体电偶极矩分别为0.23D和0.28D;在FAPbI3体系中Pb—I键长的变化范围分别为0.086 nm(GGA)和0.072 nm(LDA),其对应的PbI6八面体电偶极矩分别为0.32D和0.29D,较高的电偶极矩有利于光生电子和空穴的分离.
随后对MAPbI3和FAPbI3结构中MA+和FA+阳离子的取向展开了研究.发现当MA+阳离子中C—N键的方向平行于xoy平面,取向如图2(c)所示时,MAPbI3具有最低的能量.当FA+阳离子中N—C—N键分子所在平面垂直于xoy平面,取向如图2(d)所示时,FAPbI3具有最低的能量.MAPbI3和FAPbI3结构中MA+和FA+阳离子取向及转动引起体系总能量的变化小于0.82 eV和0.97 eV,这表明有机阳离子与周围骨架之间的静电作用并不是很强,因此有机阳离子的取向容易受到制备条件和外界环境的影响,导致其结构不稳定而容易发生相变.

表1 采用GGA和LDA方法对APbI3(A为Cs+,NH+4,MA+,FA+)进行结构优化后的晶格常数、禁带宽度、Pb—I键长,以及H和I的近邻数目Table 1.The calculated crystal parameters,band gap,Pb—I bond length and the nearest neighbor number of H and I atoms for perovskite APbI3(A denotes Cs+,NH+4,MA+,FA+)optimized with GGA and LDA.
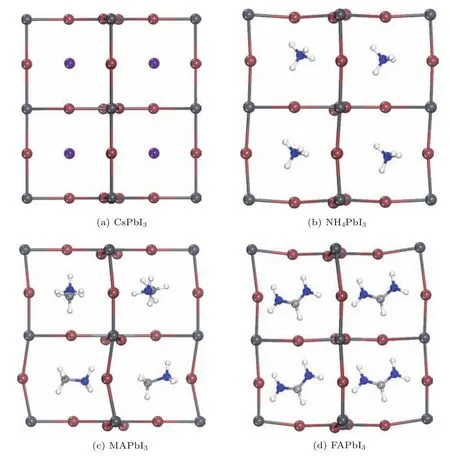
图2 (网刊彩色)采用GGA方法进行结构优化后APbI3(A为Cs+,NH,MA+,FA+)的晶体结构,其中紫色球为Cs原子,蓝色球为N原子,白色球为H原子,浅灰色球为C原子,深灰色为Pb原子,褐色球为I原子Fig.2.(color online)The geometrical structures of perovskite APbI3(A denotes Cs+,NH,MA+,FA+)optimized with GGA.Purple balls are Cs atoms,blue balls are N atoms,white balls are H atoms,light gray balls are C atoms,dark gray balls are Pb atoms,and brown balls are I atoms.
3.2 电子结构
材料的电子结构决定光生载流子的产生、复合及输运等性质.采用APbI3(A为Cs+,NH+4,MA+,FA+)全局结构优化后的几何结构进行能带结构和态密度计算.由于GGA方法计算的晶格常数更接近实验值,后续所有的能带结构和光学性质计算均采用GGA方法.表1列出了采用GGA和LDA方法获得的禁带宽度,总体上加入了OBS色散修正的LDA方法获得的禁带宽度小于加入了TS色散修正的GGA方法,两种方法计算得到的禁带宽度大小顺序是一致的.由此可见,A位置阳离子种类对其电子结构的影响较大,计算结果表明晶胞体积越小,禁带宽度越大.例如两种方法优化后的NH4PbI3具有最小的晶胞尺寸,却具有最大的禁带宽度,比FAPbI3大约0.1 eV,因而其可见光的吸收范围稍小.而CsPbI3的禁带宽度分别为1.52 eV(GGA)和0.96 eV(LDA),与其他体系相比均为最小.
四种钙钛矿材料的能带结构如图3所示,从图中可以看出,四种钙钛矿材料具有相似的能带结构,其中CsPbI3和FAPbI3均为高对称G(0,0,0)点的直接带隙半导体,禁带宽度分别为1.52 eV和1.72 eV,CsPbI3的禁带宽度计算值与实验值相当符合,仅相差0.05 eV[28],FAPbI3的禁带宽度计算值与实验值相差0.25 eV[32].而NH4PbI3和MAPbI3表现为间接带隙半导体,禁带宽度分别为1.83和1.79 eV,他们的导带底在G点左侧,能量值均比G点能量值高0.03 eV,价带顶分别在高对称点Z(0,0,0.5)和G(0,0,0)处,其中MAPbI3禁带宽度计算值与实验值比较符合(表1).由于这些能量值相差不大,可近似地认为NH4PbI3和MAPbI3是在G点的直接带隙半导体.这里计算的MAPbI3和FAPbI3禁带宽度与Lee等[33]获得的结果非常接近,差值分别为0.098和0.099 eV.这可能是由于在计算过程中没有考虑自旋耦合等因素,但是这并不影响对其电子结构的分析.
为了进一步深入分析APbI3(A为Cs+,NH+4,MA+,FA+)的电子结构,分别绘出了他们的态密度、最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)图,如图4所示.可以看出,所有体系的导带底由Pb 6p轨道和部分I 5p轨道杂化而成,价带顶由I 5p轨道组成.图4中LUMO和HOMO图更直观地显示了这些导带底和价带顶附近的轨道分布.可以看出钙钛矿结构中Cs+和FA+对价带顶附近的贡献比NH+4和MA+的贡献稍大,其中CsPbI3在其价带顶附近(−1.13 eV)存在一个较弱的态密度峰,而FAPbI3在其价带顶和导带底附近(−0.60 eV和3.31 eV)有明显的态密度峰.总体上来说,A位阳离子对费米能级附近能带的直接贡献比较弱,仅起到电荷平衡的作用.
钙钛矿材料具有优异的电荷传输能力,为此计算了APbI3(A为Cs+,NH+4,MA+,FA+)光生载流子的有效质量.载流子的有效质量为

式中a=∂2E(k)/∂k2,为能带结构中E(k)的二次函数方程中的二次项常系数,h为普朗克常数.通过第一性原理计算,获得导带底和价带顶的E(k)和k的关系,之后在其极值点附近用二次函数方程进行拟合,即可得到二次项常系数a.拟合曲线(虚线)如图3所示,其中,Eg为禁带宽度,ae和ah分别为导带底最低点和价带顶最高点二次函数方程拟合的二次项系数.再通过(1)式求出光生载流子的有效质量.从图4可以看出,CsPbI3和FAPbI3体系中导带底和价带顶拟合得到的二次项常系数a比NH4PbI3和MAPbI3大很多,即电子和空穴的有效质量较小,受外场的影响较大.
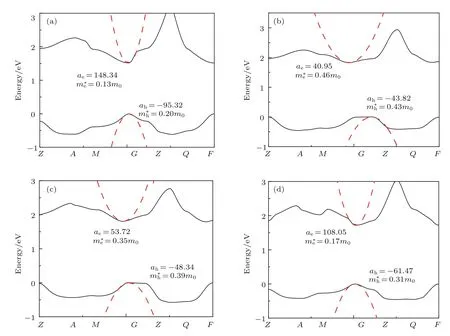
图3 钙钛矿材料APbI3(A为Cs+,NH,MA+,FA+)导带底和价带顶的能带曲线(实线),以及导带底的最低位置和价带顶最高位置附近的二次函数拟合曲线(虚线) (a)CsPbI3,Eg=1.52 eV,µ=0.08;(b)NH4PbI3,Eg=1.83 eV,µ=0.22;(c)CH3NH3PbI3,Eg=1.79 eV,µ=0.18;(d)CH(NH2)2PbI3,Eg=1.72 eV,µ=0.11Fig.3.The top curves of valence band and the bottom curves of conduction band for perovskite APbI3(A denotes Cs+,NH,MA+,FA+)(solid lines),and quadratic fitting curves(dash lines)at bottom point of conduction band and top point of valence band:(a)CsPbI3,Eg=1.52 eV,µ=0.08;(b)NH4PbI3,Eg=1.83 eV,µ=0.22;(c)CH3NH3PbI3,Eg=1.79 eV,µ=0.18;(d)CH(NH2)2PbI3,Eg=1.72 eV,µ=0.11.
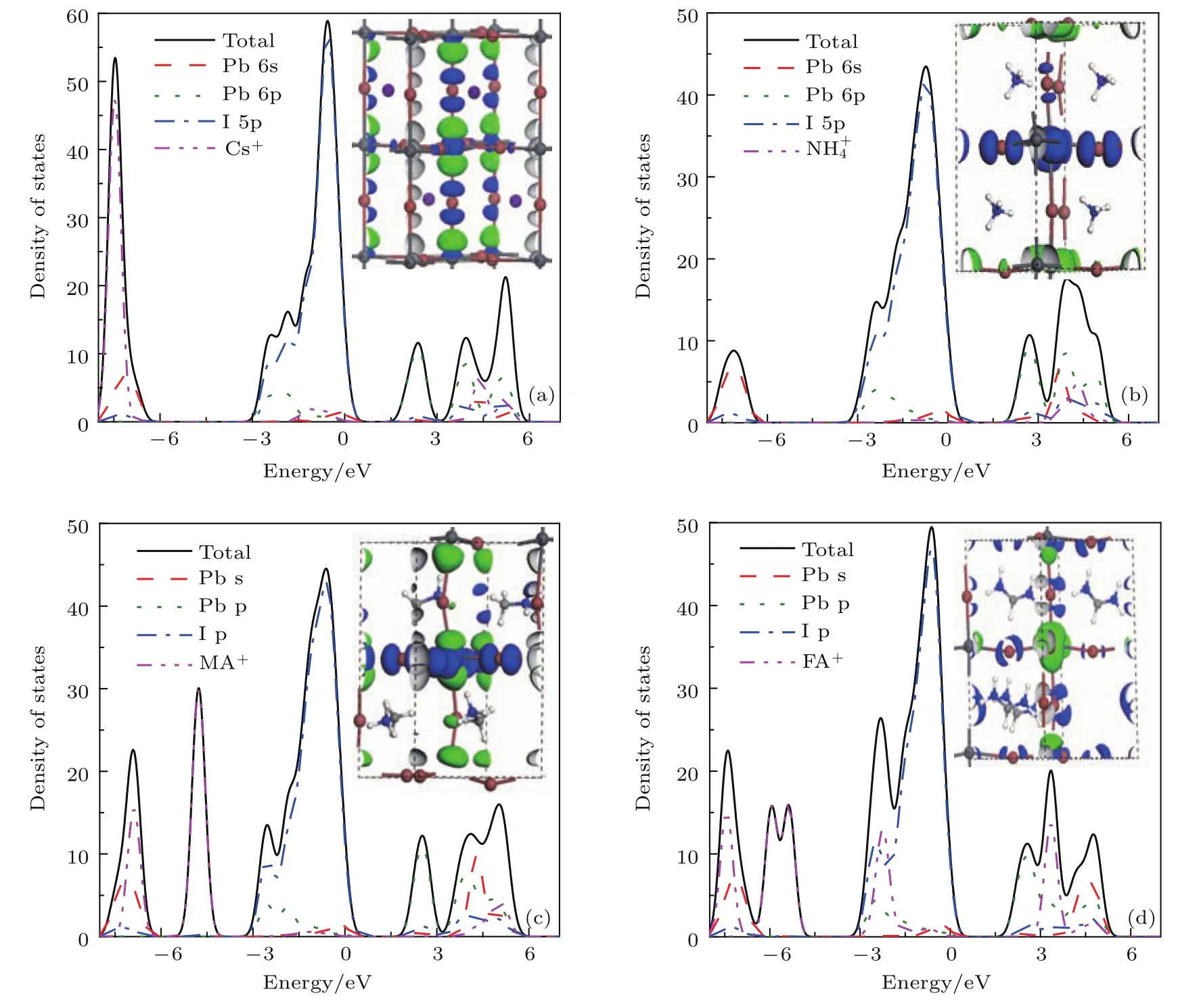
图4 (网刊彩色)采用GGA方法计算得到的APbI3(A为Cs+,NH,MA+,FA+)的总态密度和分态密度,内嵌图为LUMO图(绿色)和HOMO图(蓝色) (a)CsPbI3;(b)NH4PbI3;(c)CH3NH3PbI3;(d)CH(NH2)2PbI3Fig.4.(color online)The total and partial density of states of perovskite APbI3(A denotes Cs+,NH,MA+,FA+).The insets are LUMO(green part)and HOMO(blue part):(a)CsPbI3;(b)NH4PbI3;(c)CH3NH3PbI3;(d)CH(NH2)2PbI3.
3.3 光学性质
光学性质是半导体物理性质最重要的方面之一.计算电子结构时无论是带间还是带内跃迁频率都远超过声子频率,而且使用的方法是单电子近似方法,故仅考虑电子激发.从量子力学的观点看,带间跃迁光吸收过程是电子在辐射电磁场微扰的作用下从低能占据态到高能未占据态之间的跃迁过程.根据费米黄金定律,介电函数虚部的计算可利用电偶极子近似进行,从直接跃迁概率的定义推导出介电函数的虚部ε2为

式中u为入射电场的极化方向量;e为电子电量;Ω为倒空间体积;ε0为真空介电常数;C,V分别表示导带和价带;K为倒格矢;为动量跃迁矩阵;分别为导带和价带上的本征能级.根据Kramers-Kronig变换,可以利用介电函数的虚部得到其实部.利用吸收系数η与复折射率虚部k之间的关系可以得到吸收光谱η=2kω/c,其中c为真空中的光速,ω为圆频率.后文将采用第一性原理的计算方法获得晶体材料的吸收光谱等信息.
APbI3特殊的晶体结构使其在可见光区域内可能具有优异的光吸收特性.图5为采用GGA方法计算得到的APbI3(A为Cs+,MA+,FA+)的光吸收谱.可以看出,这四种钙钛矿材料在E//x和E//y方向存在两个较明显的光吸收峰,而且这两个方向的光吸收曲线几乎重合,但是与E//z方向光吸收峰明显不同.在CsPbI3和FAPbI3体系中,E//x,E//y和E//z的光吸收峰均处于相同的波长位置,仅光吸收系数的峰高不相同.而在NH4PbI3和MAPbI3体系中光吸收峰的位置有明显的差异.总体说来,这四种钙钛矿材料的光吸收都表现出一定的各向异性,这是由晶体结构的不对称性决定的.
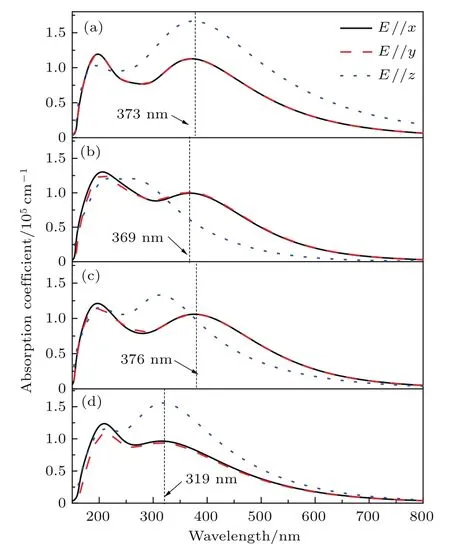
图5 (网刊彩色)钙钛矿材料APbI3(A为Cs+,NH,MA+,FA+)的光吸收谱 (a)CsPbI3;(b)NH4PbI3;(c)MAPbI3;(d)FAPbI3Fig.5. (color online)Absorption spectra of perovskite APbI3(A denotes Cs+,NH,MA+,FA+):(a)CsPbI3;(b)NH4PbI3;(c)MAPbI3;(d)FAPbI3.
在CsPbI3,NH4 PbI3,MAPbI3和FAPbI3四种体系中最大波长的吸收峰分别位于373 nm(E//x,E//y和E//z),369 nm(E//x,E//y),376 nm(E//x,E//y)和319 nm(E//x,E//y和E//z).以MAPbI3为例,计算出的光吸收峰与通过紫外-可见光谱仪测出的吸收峰390 nm情形非常接近[21].进一步观察光吸收谱可以发现,在CsPbI3和FAPbI3体系中E//z方向的光吸收峰非常高,与E//x和E//y方向相比,其光吸收边向长波方向有较大的扩展.CsPbI3的吸收带边在850 nm附近,而NH4PbI3,MAPbI3和FAPbI3的吸收带边分别位于670,690,720 nm附近,这都是由其自身的禁带宽度决定的,对应着吸收光子的电子从价带顶跃迁到导带底的电子跃迁过程.总体来说,CsPbI3体系在长波范围内的可见光和近红外区域均有较强的光吸收能力.
4 结 论
采用平面波超软赝势方法研究了APbI3(A为Cs+,NH,MA+,FA+)结构的稳定性、电子结构及光学性质.考虑到A位有机阳离子与PbI基质之间的相互作用较弱,计算时引入了相应的色散修正方案.结果显示受阳离子种类和尺寸的影响,PbI基体骨架发生不同程度的扭曲,MAPbI3和FAPbI3中PbI6八面体显示出较大的电偶极矩,这有利于光生电子和空穴的分离.四种体系中费米能级附近的能带比较相似,即价带顶均由I 5p轨道组成,导带底由Pb 6p轨道和部分I 5p轨道杂化而成.电子结构和光学性质的差异与PbI6八面体结构的扭曲程度密切相关.通过比较可以发现,CsPbI3显示出最窄的直接带隙、最小载流子有效质量和较强的光吸收能力.由于CsPbI3表现出优异的性质,将在随后的研究工作中进一步探讨混合钙钛矿CsxA1−xPbI3(A为NH+4,MA+,FA+)体系的稳定性和物理性质.
[1]Mei A Y,Li X,Liu L F,Ku Z L,Liu T F,Rong Y G,Xu M,Hu M,Chen J Z,Yang Y,Grätzel M,Han H W 2014Science345 295
[2]Kim H S,Lee C R,Im J H,Lee K B,Moehl T,Marchioro A,Moon S J,Baker R H,Yum J H,Moser J E,Grätzel M,Park N G 2012Sci.Rep.2 591
[3]Wang F Z,Tan Z A,Dai S Y,Li Y F 2015Acta Phys.Sin.64 038401(in Chinese)[王福芝,谭占鳌,戴松元,李永舫2015物理学报64 038401]
[4]Kojima A,Teshima K,Shirai Y,Miyasaka T 2009J.Am.Chem.Soc.131 6050
[5]Yang W S,Noh J H,Jeon N J,Kim Y C,Ryu S,Seo J,Seok S II 2015Science348 1234
[6]Zhang D F,Zheng L L,Ma Y Z,Wang S F,Bian Z Q,Huang C H,Gong Q H,Xiao L X 2015Acta Phys.Sin.64 038803(in Chinese)[张丹霏,郑灵灵,马英壮,王树峰,卞祖强,黄春辉,龚旗煌,肖立新2015物理学报64 038803]
[7]Cappel U B,Daeneke T,Bach U 2012Nano Lett.12 4925
[8]Liu M Z,Johnston M B,Snaith H J 2013Nature501 395
[9]Knop O,Wasylishen R E,White M A,Oort M J M V 1990Can.J.Chem.68 412
[10]Lee J W,Seol D J,Cho A N 2014Adv.Mater.26 4991
[11]Zhou Y Y,Yang M J,Pang S P,Zhu K,Padture N P 2016J.Am.Chem.Soc.138 5535
[12]Pang S P,Hu H,Zhang J L,Lv S L,Yu Y M,Wei F,Qin T S,Xu H X,Liu Z L,Cui G L 2014Chem.Mater.26 1485
[13]Choi H,Jeong J,Kim H B,Kim S,Walker B,Kim G H,Kim J Y 2014Nano Energy7 80
[14]Saliba M,Matsui T,Seo J Y,Domanski K,Correa-Baena J P,Nazeeruddin M K,Zakeeruddin S M,Tress W,Abate A,Hagfeldt A,Grätzel M 2016Energy Environ.Sci.9 1989
[15]Baikie T,Fang Y A,Kadro J M,Schreyer M,Wei F X,Mhaisalkar S G,Grätzel M,White T J 2013J.Mater.Chem.A1 5628
[16]Motta C,Mellouhi F E,Kais S,Tabet N,Alharbi F,Sanvito S 2015Nat.Commun.6 7026
[17]Filippetti A,Mattoni A 2014Phys.Rev.B89 12503
[18]Mosconi E,Amat A,Nazeeruddin M K,Grätzel M,De Angelis F 2013J.Phys.Chem.C117 13902
[19]Geng W,Zhang L,Zhang Y N,Lau W M,Liu L M 2014J.Phys.Chem.C118 19565
[20]Wang Y,Gould T,Dobson J F,Zhang H M,Yang H G,Yao X D,Zhao H J 2014J.Phys.Chem.Chem.Phys.16 1424
[21]Umari P,Mosconi E,De Angelis F 2014Sci.Rep.4 4467
[22]Kawamura Y,Mashiyama H,Hasebe K 2002J.Phys.Soc.Jpn.71 1694
[23]Vanderbilt D 1990Phys.Rev.B41 7892
[24]Tkatchenko A,Scheffler M 2009Phys.Rev.Lett.102 073005
[25]Ortmann F,Bechstedt F,Schmidt W G 2006Phys.Rev.B73 205101
[26]Monkhorst H J,Pack J D 1976Phys.Rev.B13 5188
[27]Segall M D,Lindan P J D,Probert M J,Pickard C J,Hasnip P J,Clark S J,Payne M C 2002J.Phys:Condens.Matter.14 2717
[28]Chung L,Lee B,He J Q,Chang R P H,Kanatzidis M G 2012Nature485 486
[29]Gao X,Uehara K,Klug D D,Patchkovskii S,Tse J S,Tritt T M 2005Phys.Rev.B72 125202
[30]Tanaka K,Takahashi T,Ban T,Kondo T,Uchida K,Miura N 2003Solid State Commun.127 619
[31]Schulz P E,Edri E,Kirmayer S,Hodes G,Cahen D,Kahn A 2014Energy Environ.Sci.7 1377
[32]Jeon N J,Noh J H,Yang W S,Kim Y C,Ryu S 2015Nature517 476
[33]Lee C,Hong J,Stroppa A,Whangbo M H,Shim J H 2015RSC Advances5 78701
PACS:71.15.Mb,71.20.–b,81.07.Pr,88.40.H– DOI:10.7498/aps.66.057103
Theoretical study on the stability and photoelectric properties of APbI3perovskite∗
Liu Na1)Wei Yang1)Ma Xin-Guo1)2)†Zhu Lin1)Xu Guo-Wang1)2)Chu Liang3)Huang Chu-Yun1)2)‡
1)(School of Science,Hubei University of Technology,Wuhan 430068,China)
2)(Hubei Collaborative Innovation Center for High-Efficiency Utilization of Solar Energy,Hubei University of Technology,Wuhan 430068,China)
3)(School of Science,Nanjing University of Posts and Telecommunications,Nanjing 210046,China)
29 July 2016;revised manuscript
4 December 2016)
The rapid development of organic-inorganic hybrid perovskite solar cells has recently attracted the worldwide attention because their power conversion efficiency has risen from 4%to higher than 20%within just six years.It is well known that the perovskite materials with APbI3crystal structure have a 3D framework of corner-sharing PbI6octahedra,in which each Pb atom bonds with six I atoms,and the A cations fill in the octahedral interstices.At present,a lot of researches have focused on the synthesis and doping modification of perovskite materials.However,it is hard to detect directly the weak interactions between A cations and PbI6skeleton in the APbI3crystal structure through experiments,which have effect on the structural stability and electronic properties.To provide a full understanding of the interplay among size,structure,and organic/inorganic interactions,the stability,electronic structures and optical properties of APbI3were investigated by the plane-wave ultra soft pseudo potentials.Two dispersion corrections were taken into account in the weak interactions between A cations and PbI6skeleton in the APbI3crystal structure,respectively.The results show that the type and size of cations affect the distortion of PbI framework,indicating that the larger the radius of the A cation is,the stronger the interaction between the A cation and the PbI framework is.Further,it is identified that after geometry relaxation,the orientation of A cations(A denotesis easy to change,and the PbI frameworks present structural distortion.CsPbI3is more stable energetically than other three kinds of perovskite materials.For the PbI6octahedra,the large dipole moments of 0.23D and 0.32D for the generalized-gradient approximation method or 0.28D and 0.29D for the local-density approximation method are also present in MAPbI3and FAPbI3,respectively.In addition,the energy band structures,which affect the generation and migration of photon-generated carriers and optical properties,will alter with the structural distortion of PbI frameworks.By analyzing the energy band structures and corresponding density of states,we find that four systems have similar band structures near the Fermi energy,namely,the top of valance band is mainly contributed by I 5p orbitals,while the bottom of conduction band is dominated by Pb 6p orbitals and partly contributed by I 5p orbitals.A little difference of their electronic structures and optical absorption spectra originates from the distortion of PbI6octahedra in APbI3crystal structures.It is noted that the contribution of the ions Cs+and FA+on the top of valance band is slightly larger than that of the ions NH+4and MA+.Compared with other three kinds of perovskite materials,CsPbI3presents the narrowest direct band gap,the lowest effective carrier mass and excellent visible-light and infrared absorption.The results may provide some theoretical guidance for further research on perovskite materials in the application of solar cells.
perovskite material,structural stability,electronic structure,first-principles
PACS:71.15.Mb,71.20.–b,81.07.Pr,88.40.H–
10.7498/aps.66.057103
∗国家自然科学基金(批准号:51472081)、湖北工业大学高层次人才启动基金(批准号:GCRC13014)、绿色工业引领计划(批准号:YXQN2016005)和湖北省协同创新中心开放基金(批准号:HBSKFZD2014003,HBSKFZD2014011,HBSKFZD2015004)资助的课题.
†通信作者.E-mail:maxg2013@sohu.com
‡通信作者.E-mail:chuyunh@163.com
*Project supported by the National Natural Science Foundation of China(Grant No.51472081),the Foundation of Hubei University of Technology for High-Level Talents(Grant No.GCRC13014),the Leading Plan of Green Industry(Grant No.YXQN2016005),and the Development Founds of Hubei Collaborative Innovation Center(Grant Nos.HBSKFZD2014003,HBSKFZD2014011,HBSKFZD2015004).
†Corresponding author.E-mail:maxg2013@sohu.com
‡Corresponding author.E-mail:chuyunh@163.com
