Ti3AC2相(A=Si,Sn,Al,Ge)电子结构、弹性性质的第一性原理研究∗
2017-08-01胡洁琼谢明陈家林刘满门陈永泰王松王塞北李爱坤
胡洁琼 谢明 陈家林 刘满门 陈永泰 王松 王塞北 李爱坤
(昆明贵金属研究所,稀贵金属综合利用新技术国家重点实验室,昆明 650106)
Ti3AC2相(A=Si,Sn,Al,Ge)电子结构、弹性性质的第一性原理研究∗
胡洁琼 谢明†陈家林 刘满门 陈永泰 王松 王塞北 李爱坤
(昆明贵金属研究所,稀贵金属综合利用新技术国家重点实验室,昆明 650106)
(2016年10月9日收到;2016年12月1日收到修改稿)
采用第一性原理的密度泛函理论平面波赝势法,通过广义梯度近似研究了Ti3AC2相(A=Si,Sn,Al,Ge)的相结构、能量、电子结构和弹性性质.首先对六方晶相结构的Ti3AC2(A=Si,Sn,Al,Ge)四个相进行几何优化,对其能带结构、总态密度、分态密度和电荷密度分布以及弹性性质进行研究,并计算各相的内聚能与形成能.计算结果表明:Ti3GeC2较其他三相稳定,Ti3AlC2的形成能最低,说明Ti3AlC2较Ti3SiC2,Ti3SnC2和Ti3GeC2更易生成;Ti3AC2(A=Si,Sn,Al,Ge)各相在费米能级处的电子态密度较高,材料表现出较强的金属性,同时各相的导电性为各向异性.Ti3AC2(A=Si,Sn,Al,Ge)各相的导电性主要由Ti的3d电子决定,A(A=Si,Sn,Al,Ge)的p态电子和C的2p态电子也有少量贡献.决定材料电学性质的主要是Ti的3d,A的p和C的2p态电子的p-d电子轨道杂化,而p-d电子轨道杂化成键则使材料具有比较稳定的结构;对Ti3AC2相(A=Si,Sn,Al,Ge)弹性性质的研究表明Ti3AlC2的原子间结合力较弱,而Ti3GeC2的原子间结合力相对较强,材料的强度较大.
第一性原理,MAX相,电子结构,弹性性质
1 引 言
Mn+1AXn系列三元合金化合物是层状化合物,其中M代表过渡金属元素(Ti,Zr,Cr,Mo);A代表主族元素(Al,Si,P,S,Ga,Ge,Sn);X代表碳或氮.MAX相材料的晶体结构属于六方晶系,空间群为P63/mmc,其晶体结构可以描述为由类似岩盐型结构的Mn+1Xn片层与紧密堆积的A原子层在c方向上交替堆垛而组成.根据通式中n取值的不同(即MX片层厚度的不同),MAX相可以划分为M2AX相、M3AX2相以及M4AX3相等.这种特殊的原子排列方式使MAX相材料具备了层状结构的特点,层状结构中存在着多种类型的化学键,且具有主体原子层化学组成可调,层内弹性空间可调等优点.三元层状陶瓷集金属和陶瓷的优点于一身,在常温下能保持较好的导电性、导热性和抗热震性、高温塑性、易加工性,有较高的断裂韧性和屈服强度,高熔点和高热稳定性,良好的抗氧化性和耐腐蚀性,在高温时仍具有非常好的强度,同时还有非常低的摩擦系数以及很好的自润滑性能[1−3].因此,近年来三元层状陶瓷基复合材料作为新型结构/功能材料,以其独特的性能吸引了无数材料工作者,具有很好的研究价值和非常广阔的应用前景[4−6].
Ti3SiC2的晶体结构最早由Jeitschko等[7]测定,其晶格参数为a=0.307 nm,c=1.769 nm,共棱的Ti6C八面体被平行四边形的硅原子层分隔.和Ti3AlC2材料相似,Ti3SiC2材料同样综合了金属和陶瓷的双重性能,适宜于在高温或其他氧化环境下用作轴承材料和机车的润滑材料,高温下的高屈服点和塑性,使得Ti3SiC2在高温结构方面的应用更具有优势,如作陶瓷发动机材料等.
1994年,Pietzka和Schuster[8]最早报道了合成Ti3AlC2材料,Ti3AlC2材料综合了金属和陶瓷的双重优异性能:具有良好的导电性,低硬度、高强度,高弹性模量可进行机械加工,高热稳定性和优异的抗氧化性能等.Ti3AlC2材料具有层状结构,紧密堆积的Ti6C八面体被铝原子层分隔开来,晶体中每一个单胞中包含两个Ti3AlC2分子.
Ti3GeC2最早是由Barsoum[9]合成出来的,实验上测定其晶格常数为a=0.307 nm,c=1.776 nm.Ti3GeC2除了具有金属和陶瓷的双重优异性能外,在紫外光区还具有较大且稳定的反射率,因此可作为潜在的新型防紫外及短波涂层材料以及航天器用温度控制涂层材料.
Benoit[10]通过热静压实验合成了Ti3SnC2,Ti3SnC2在结构上与Ti3SiC2和Ti3GeC2相同,因此Ti3SnC2,Ti3SiC2和Ti3GeC2在许多方面具有类似的性质.
目前关于Ti3AC2相(A=Si,Sn,Al,Ge)的电子结构理论和弹性性能的研究报道较少.而MAX相的许多性质都是由其电子结构性能决定的,因此研究其电子结构和弹性性能对探索材料特性非常有意义.本工作在密度泛函理论框架下采用第一性原理研究分析了Ti3AC2相(A=Si,Sn,Al,Ge)的电子结构、成键特性和弹性性能,为完善合成高纯Ti3AC2相(A=Si,Sn,Al,Ge),以及其他性质的深入研究和工业上的推广使用奠定理论基础.
2 计算方法与模型
本工作的计算是基于密度泛函理论的第一性原理方法,利用了美国Accelrys公司Material Studios2016软件中的量子力学模块CASTEP(Cambridge serial total energy package)软件包完成[11].CASTEP软件是一个基于密度泛函方法的从头算量子力学程序.利用总能量的平面波赝势方法,将离子势用赝势替代,电子-电子相互作用的交换和相关势由广义梯度近似(GGA)中的PBE(Perdew,Burke and Ernzerhof)[12]形式进行校正,它是目前较为准确的电子结构计算的理论方法.赝势取倒易空间表征中的超软(ultrasoft)赝势.采用周期边界条件,K点网格数取 13×13×2,晶体中电子波函数通过平面波基组展开,平面波数目由动能截断点来决定,计算晶胞所选取的能量截断值(cut-o ff)为370 eV.在进行各项计算之前都用BFGS(Broyden-Flecher-Goldfarb-Shanno)方法对晶胞进行几何优化,以求得它们的局域处于最稳定结构.自洽计算的收敛精度为 5.0×10−7eV/atom,每个原子上的力要求低于0.01 eV/atom,公差偏移小于 5.0×10−4Å,应力偏差小于0.02 GPa.
Ti3SiC2,Ti3SnC2,Ti3AlC2,Ti3GeC2的晶体结构属于六方晶系,空间群为P63/mmc.Ti3SiC2的原胞参数为a=b=3.067 Å,c=17.675 Å,α=β=90◦,γ=120◦[13];各原子的内坐标为Ti1(0,0,0),Ti2(2/3,1/3,0.1355),Si(0,0,1/4),C(1/3,2/3,0.0723).Ti3SnC2的原胞参数为a=b=3.137 Å,c=18.650 Å,α=β=90◦,γ=120◦[14];各原子的内坐标为 Ti1(0,0,0),Ti2(2/3,1/3,0.1266),Sn(0,0,1/4),C(1/3,2/3,0.0693).Ti3AlC2的原胞参数为a=b=3.073 Å,c=18.557 Å,α=β=90◦,γ=120◦[15]; 各原子的内坐标为Ti1(0,0,0),Ti2(2/3,1/3,0.1276),Al(0,0,1/4),C(1/3,2/3,0.0691).Ti3GeC2的原胞参数为a=b=3.077 Å,c=17.76 Å,α=β=90◦,γ=120◦[16];各原子的内坐标为Ti(0,0,0),Ti(2/3,1/3,0.1338),Ge(0,0,1/4),C(1/3,2/3,0.5723).它们的晶胞模型如图1所示.
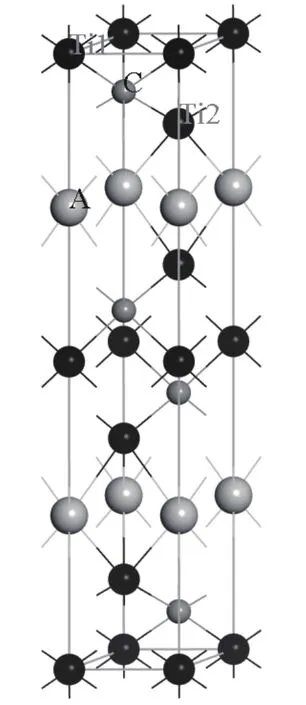
图1 Ti3AC2相的晶体结构模型Fig.1.Crystal structure of Ti3AC2(A=Si,Sn,Al,Ge)phases.
3 结果与讨论
3.1 Ti3AC2(A=Si,Sn,Al,Ge)相的内聚能
经过优化计算,获得了Ti3SiC2,Ti3SnC2,Ti3AlC2,Ti3GeC2的基态晶格结构,计算得到的优化晶格参数见表1.通过表1可以看出,GGA的理论计算值与实验值均能很好地符合,说明计算结果是可信的.
由中性原子结合成晶体所释放出的能量或将晶体拆散成中性原子所消耗的能量称为晶体的结合能或内聚能,反映了晶体的稳定性和键合强度.显然,晶体的内聚能就等于它的升华热.根据定义,可以使用下式计算MAX相的内聚能Ecoh[17]:

total 为Ti3AC2的总能,为纯M原子的赝原子能.
形成能是指由单质直接合成相化合物的过程中所降低的能量.计算公式可以表示如下:
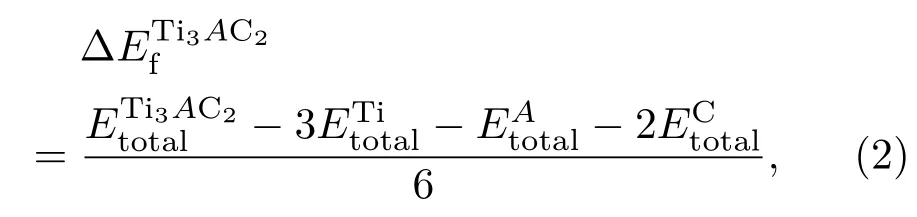
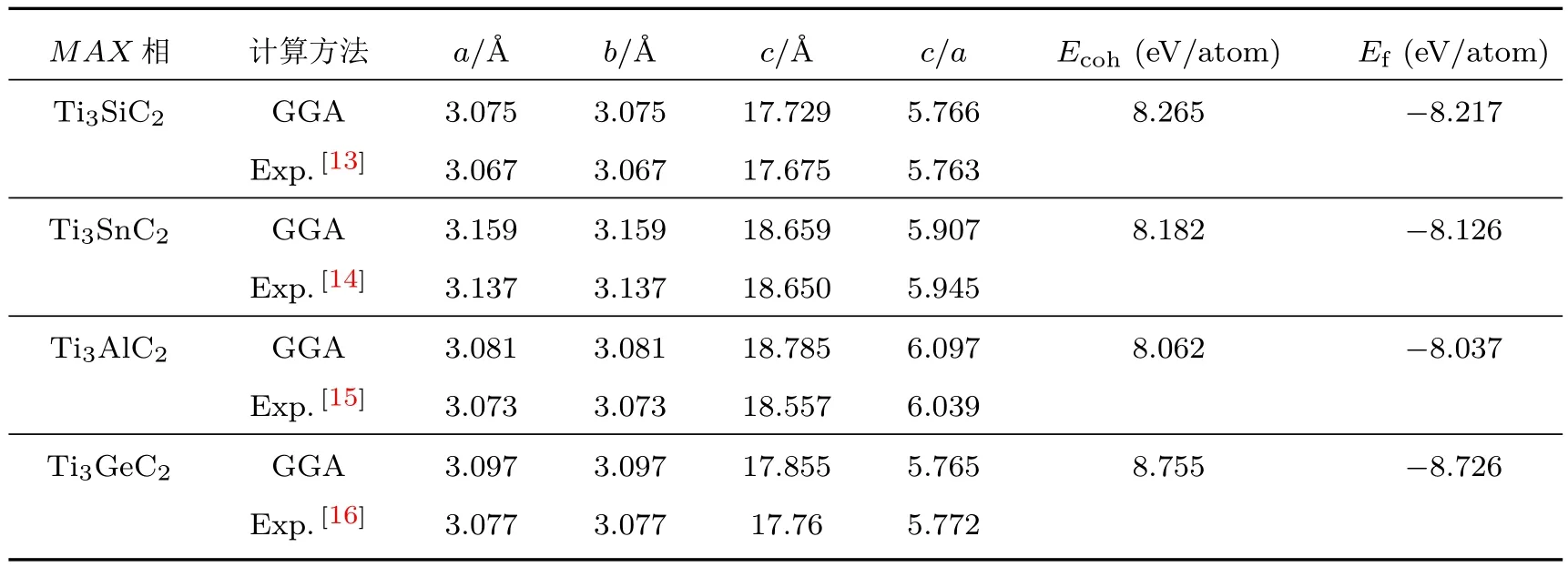
表1 Ti3AC2(A=Si,Sn,Al,Ge)相的晶胞参数、内聚能与形成能Table 1.Lattice parameter,cohesive energies and formation energies of Ti3AC2(A=Si,Sn,Al,Ge)phases.
Ti3AC2相的内聚能和形成能如表1所列.内聚能是判断材料键合力强弱的重要指标,内聚能越低其晶体键合力就越弱,内聚能越高则晶体的键合力就越强.由表1可以看出从Ti3GeC2,Ti3SiC2,Ti3SnC2到Ti3AlC2的内聚能逐渐减小,说明Ti3GeC2的键合力较强,而Ti3AlC2的键合力最弱.所研究化合物的形成能均为负值,这表明它们相对单质元素在热力学上是稳定的.Ti3GeC2的形成能最低,说明Ti3GeC2较Ti3SiC2,Ti3SnC2和Ti3AlC2更加稳定.
3.2 电子结构性质
在几何结构优化后,Ti3SiC2,Ti3SnC2,Ti3AlC2,Ti3GeC2沿 Brillouin区高对称点方向的能带结构计算结果见图2,其中能量值在0 eV位置处的垂直虚线表示费米能的位置.由图2可以看出,Ti3SiC2,Ti3SnC2,Ti3AlC2,Ti3GeC2的导电性表现出很强的金属材料的特征,图中无明显的带隙,电子很容易获得能量而跳跃至导带而导电.从能带结构图还可以看出各Ti3AC2(A=Si,Sn,Al,Ge)相分布的能量不对称,因此其导电性为各向异性.Ti3SiC2,Ti3SnC2,Ti3AlC2,Ti3GeC2的价带基本都位于−34—−33 eV和−13—0 eV两个区域,Ti3SiC2,Ti3AlC2的导带位于0—20 eV,而Ti3SnC2的导带位于0—3 eV,Ti3GeC2的导带位于0—25 eV.从图2还可更直观地看到各相能带的幅度变化和能级穿越情况.Ti3SiC2和Ti3GeC2的能带较宽,也即在能带图中的起伏较大,说明处于这个带中的电子有效质量较小,非局域的程度较大,组成这条能带的原子轨道扩展性较强.而Ti3SnC2的能带较窄,表明对应于这条能带的本征态主要是由局域于某个格点的原子轨道组成,这条带上的电子局域性非常强,有效质量相对较大.Ti3SiC2和Ti3GeC2相邻轨道之间的重叠较大,且Ti3GeC2相邻轨道之间的重叠最大,表明Ti3GeC2成键强度较强.

图2 Ti3AC2(A=Si,Sn,Al,Ge)相的能带结构图 (a)Ti3SiC2;(b)Ti3SnC2;(c)Ti3AlC2;(d)Ti3GeC2Fig.2.Band structures of Ti3AC2(A=Si,Sn,Al,Ge):(a)Ti3SiC2;(b)Ti3SnC2;(c)Ti3AlC2;(d)Ti3GeC2.
图3为Ti3SiC2,Ti3SnC2,Ti3AlC2,Ti3GeC2的总态密度图和分态密度图.通过对分态密度图的分析,可以推断出组成化合物的各原子间的成键情况.费米能级以上,Ti的3d电子占据主导地位,而Si,Sn,Al,Ge的p和C的2p电子有少量贡献;费米能级以下,Ti的3d轨道电子、C的2p轨道电子和Si,Sn,Al,Ge的p轨道电子相互之间形成了较强的共价键.从图3(a)Ti3SiC2的态密度图可以看出,Si的3s电子主要位于−11.5—−6.0 eV能量区间,少量位于−5.2—5.0 eV能量区间;而Si的3p电子则主要位于−7.5—5.0 eV能量区间,少量位于−11.5—−6.0 eV能量区间,因此Si的3s和3p电子轨道在−11.5—5.0 eV能量区间相互重叠,电子轨道发生杂化.对比图3(a)中Si和C的态密度图可以看出,Si的3s电子态占据的宽度远大于C的2s电子态,Si的3s电子的态密度峰数量多于C的2s态,表明sp杂化主要为Si的3s电子和3p电子之间的杂化,而Si的3p态电子则和C的2p态电子轨道的杂化程度较高.在−12.5—−10.0 eV能量区间,Si的3s轨道电子与C的2s轨道电子形成的Si—C键占主导地位,Si-C的相互作用对结构的稳定性也起着重要作用.在−7—−2.0 eV能量区间,Si的3p轨道电子和C的2p轨道电子分别与Ti的3d轨道电子形成的p—d键占主导地位.从图3(a)中还可看出Ti的3d电子主要位于−5—5.0 eV能量区间,因此Si的3s电子和3p电子与Ti的3d电子之间存在p-d和sp-d形式的轨道杂化.Ti的3d轨道电子和Si的3p轨道电子主要在费米能级下的−4.8—−1.2 eV能量区间成键,而p轨道电子和d轨道电子杂化形成的p—d键使Ti3SiC2具有稳定的结构.Ti的3d电子在费米能级处的态密度峰值为2.8,明显高于Si和C的电子态密度峰值,因此Ti3SiC2的高电导率主要是由费米能级附近Ti的3d态电子主导.从图3(b)中可以看出,Sn的5s电子主要位于−11.0—−5.0 eV能量区间,少量位于−5.2—3.0 eV能量区间;而Sn的5p电子则主要位于−6.2—2.0 eV能量区间,少量位于−10.0—−6.0 eV能量区间.因此Sn的5s和5p电子轨道在−11.0—−7.5 eV能量区间相互重叠,电子轨道发生杂化.Sn的5s电子态占据的宽度远大于C的2s电子态,Sn的5s电子的态密度峰数量多于C的2s态,表明sp杂化主要为Sn的5s电子和5p电子之间的杂化,而Sn的5p态电子则和C的2p态电子轨道的杂化程度较高.在−12.0—−10 eV能量区间,Sn的5s轨道电子与C的2s轨道电子形成的Sn—C键占主导地位,Sn—C的相互作用对结构的稳定性也起着重要作用.在−6—−0.6 eV能量区间,Sn的5p轨道电子和C的2p轨道电子分别与Ti的3d轨道电子形成的p—d键占主导地位.图3(d)中Ti3GeC2的成键特点与Ti3SiC2相似,只是除了Ge的sp杂化外,在7—17.0 eV的能量区间内还存在Ge的4s与Ti的3p的sp杂化.

图3 Ti3AC2(A=Si,Sn,Al,Ge)相的总态密度图和分态密度图 (a)Ti3SiC2;(b)Ti3SnC2;(c)Ti3AlC2;(d)Ti3GeC2Fig.3.Total and partial density of states of Ti3AC2(A=Si,Sn,Al,Ge):(a)Ti3SiC2;(b)Ti3SnC2;(c)Ti3AlC2;(d)Ti3GeC2.
从图3(c)中可以看出,Al的3s电子主要位于−8.5—−3.0 eV能量区间,少量位于0—5.0 eV能量区间;而Al的3p电子则主要位于−7—7.0 eV能量区间,少量位于10—15.0 eV能量区间,因此Al的3s和3p电子轨道在0—5.0 eV能量区间相互重叠,电子轨道发生杂化.对比图3(c)中Al和C的态密度图可以看出,Al的3s电子态占据的宽度远大于C的2s电子态,Al的3s电子的态密度峰数量多于C的2s态,表明sp杂化主要为Al的3s电子和3p电子之间的杂化,而Al的3p态电子则和C的2p态电子轨道的杂化程度较高.在0—7.0 eV能量区间Al的3p轨道电子和C的2p轨道电子分别与Ti的3d轨道电子形成的p—d键占主导地位.从图3(c)中还可看出Ti的3d电子主要位于−5—5.0 eV能量区间,因此Al的3s电子和3p电子与Ti的3d电子之间存在p-d和sp-d形式的轨道杂化.Ti的3d轨道电子和Al的3p轨道电子主要在费米能级下的−5—−1.2 eV能量区间成键,Ti的3d轨道电子和C的2p轨道电子主要在费米能级下的−7—−2 eV能量区间成键,而p轨道电子和d轨道电子杂化形成的p—d键使Ti3AlC2具有稳定的结构.
表2中列出了Ti3AC2相中各种成键形式以及各键的键长和各相在费米能级处的态密度值.一般影响键长大小的因素主要是成键两原子的性质、体积及化学键类型.通常,键能越大键长越短,成键原子间化学键数目越多则键长越短,键长越短,键能越大,材料越稳定[18].由于Ti3AC2相材料是层状结构化合物,因此Ti-Si/Sn/Al/Ge之间存在着很强的金属键,Ti—C之间则作用着极强的共价键.从表2中可以看出三种相的Ti—C键长基本一致,不同的是Ti—Si/Sn/Al/Ge键,其中Ti3GeC2中的Ti—Ge键键长最短,说明Ti—Ge键键能较大,这与之前分析态密度时提到的Ti,Ge的电子轨道之间存在sp杂化有关.费米能级的位置和费米能级处态密度的数N(EF)决定着材料的稳定性,费米能级处态密度的数值N(EF)越低,材料结构越稳定.由于Ti3GeC2的费米能级处态密度N(EF)值相对较低,说明Ti3GeC2的稳定性相对较高.

表2 Ti3AC2(A=Si,Sn,Al,Ge)相中各键的键长和各相在费米能级处的态密度值Table 2.Bond length(Å)and density of states at Femi level(eV−1)of Ti3AC2(A=Si,Sn,Al,Ge)
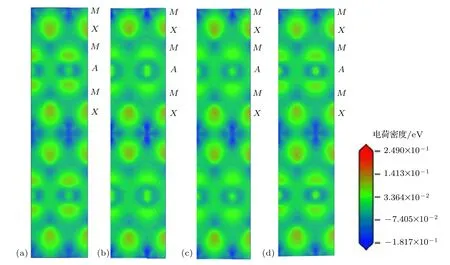
图4 (网刊彩色)Ti3AC2(A=Si,Sn,Al,Ge)(100)面的电荷密度图 (a)Ti3SiC2;(b)Ti3SnC2;(c)Ti3AlC2;(d)Ti3GeC2Fig.4.(color online)Charge densities on(100)plane of(a)Ti3SiC2,(b)Ti3SnC2,(c)Ti3AlC2,(d)Ti3GeC2.
为了更加直观形象地理解Ti3AC2(A=Si,Sn,Al,Ge)各相中原子间的成键情况,选取各个晶胞(100)面的电荷密度分布图进行分析,如图4所示.由图4可以看出,Ti3AC2晶胞中,M-X之间的电荷密度较高,而M-A和A-X之间的电荷密度较低,故M-X之间存在较强的相互作用,而M-A和A-X之间的作用不明显;比较Ti3AC2各晶胞的电荷密度图可以看出,各晶胞电荷密度分布的主要差别在于M-A之间相互作用的强弱,由图4可以看出M-A之间的键合由强到弱的顺序为:Ti3GeC2和Ti3SiC2中M-A之间的电荷密度较大,键合强度较高,而Ti3SnC2和Ti3AlC2中M-A之间的电荷密度较小,键合强度较小.
3.3 弹性性质
为了了解Ti3AC2相材料的力学性能,本文用优化后的完整晶胞计算了Ti3AC2相材料的弹性常数和弹性模量,弹性常数反映了晶体结构的力学稳定性,而弹性模量则反映了材料对弹性变形的抗力,其中弹性模量包括杨氏模量、体积弹性模量、剪切弹性模量等.由于Ti3AC2相材料属于六方晶系,计算出来的弹性常数可以用来判断Ti3AC2相材料的力学稳定性.波恩和黄昆提出的六方晶系的力学稳定性需要满足以下关系[21]:

本文计算了Ti3SiC2,Ti3SnC2,Ti3AlC2和Ti3GeC2这四种材料的弹性常数和弹性模量,如表3所列.通过CASTEP软件计算,Ti3SiC2的弹性常数C11=307>0,C11−C12=210>0,C44=116>0,C66=105>0,(C11+C12)C33=满足(1)式的判定条件,有力地证明了Ti3SiC2能够稳定存在且具有力学稳定性.同理,Ti3SnC2的弹性常数C11=324>0,C11−C12=235>0,C44=110>0,C66=118>0,13122. Ti3AlC2的弹性常数C11=269>0,C11−C12=172>0,C44=78>0,C66=86>0,Ti3GeC2的弹性常数C11=320>0,C11−C12=217>0,C44=123>0,C66=108>0,以上数据表明Ti3SnC2,Ti3AlC2和Ti3GeC2材料也都是能够稳定存在且具有力学稳定性的.
弹性常数还可以说明MAX相材料的弹性各向异性问题.具有六方晶体结构的MAX相材料的各向异性通常用以下三种参数来判定[22]:
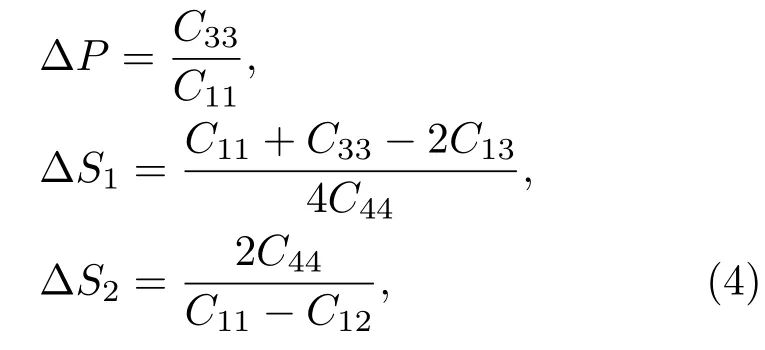
其中,∆P表示晶体的压缩波的各向异性情况,∆S1和∆S2表示剪切波的各向异性情况.由于他们的值都不为1,说明晶体的弹性是各向异性的,否则就为各向同性.
通过计算得到的弹性常数,采用Vogit-Reuss-Hill近似[23]计算MAX相材料的弹性模量,Hill近似是Vogit近似和Reuss近似的简单算数平均数,计算得到的弹性模量有体模量、剪切模量和杨氏模量,计算结果也在表3中列出.弹性模量表明了材料对弹性形变的抵抗能力,即材料变形的难易程度.弹性模量的值越大,在相同的应力作用下,材料的弹性形变就越小,受到外力作用时,材料保持其固有尺寸和形状的能力就越强.表3中所示的Ti3SiC2材料的体模量B和剪切模量G的值分别为183和100 GPa,G/B=0.546.由于0.546<0.57,说明Ti3SiC2材料是韧性的.同理,可以计算得到的Ti3AlC2,Ti3SnC2和Ti3GeC2材料的G/B值都大于0.57,说明Ti3AlC2,Ti3SnC2和Ti3GeC2材料是偏脆性的.
体模量是晶体抗体积变化能力的量度,可以反映材料抵抗断裂的能力[24].从表3可以看出Ti3SiC2有很强的抗体积变化的特性.剪切模量是晶体抗形状变化能力的量度,可以反映抵抗塑性变形的能力,一般来说,很高的剪切模量值意味着原子间很明显的方向价键特性[25].计算结果表明,Ti3GeC2中的原子价键是最强的.弹性模量是表征在弹性限度内物质材料抗拉或抗压的物理量,它是沿纵向的弹性模量一种最重要、最具特征的力学性质[26].弹性模量可视为衡量材料产生弹性形变难易程度的指标,其值越大,使材料发生一定弹性形变的应力也越大,即材料的强度越大,亦即在一定应力作用下,发生弹性形变越小.从原子间相互作用力的角度来看,弹性模量值的大小反映了原子间结合力的大小.Ti3AlC2的弹性模量的值相对较小,初步可以判断其原子间结合力较弱.而Ti3GeC2材料则有较高的弹性模量,说明其原子间结合力相对较强,材料的强度较大.

表3 Ti3AC2(A=Si,Sn,Al,Ge)相材料的弹性常数、体模量、剪切模量和弹性模量Table 3.Elastic constants,bulk modules,shear modules and elasticity modulus for Ti3AC2(A=Si,Sn,Al,Ge)phases.
4 结 论
1)计算得到的Ti3AC2(A=Si,Sn,Al,Ge)各相的平衡晶格常数与实验值十分符合,取得了可信的晶体模型.通过内聚能和形成能的计算和比较,得出从Ti3GeC2,Ti3SiC2,Ti3SnC2到Ti3AlC2的内聚能逐渐减小,说明Ti3GeC2的键合力较强,而Ti3AlC2的键合力最弱.Ti3GeC2的形成能最低,说明Ti3GeC2较Ti3SiC2,Ti3SnC2和Ti3AlC2更加稳定.
2)从电子结构的角度分析各Ti3AC2(A=Si,Sn,Al,Ge)材料得出,在费米能级上,Ti的3d电子占据主导地位,而Si,Sn,Al,Ge的p和C的2p电子有少量贡献;费米能级以下,Ti的3d轨道电子、C的2p轨道电子和Si,Sn,Al,Ge的p轨道电子相互之间形成了较强的共价键.决定Ti3AC2(A=Si,Sn,Al,Ge)材料电学性质的主要是Ti的3d,A的p和C的2p态电子的p-d电子轨道杂化;Ti3SiC2,Ti3SnC2和Ti3AlC2具有类似的电子杂化特性,而Ti3GeC2除了Ge的sp杂化外,还存在Ge的4s与Ti的3p的sp杂化.
3)通过对Ti3AC2(A=Si,Sn,Al,Ge)材料弹性性质的计算可以得出,Ti3AC2(A=Si,Sn,Al,Ge)材料都是能够稳定存在且具有力学稳定性的,且其弹性都是各向异性的;Ti3SiC2材料是韧性的,而Ti3AlC2,Ti3SnC2和Ti3GeC2材料是偏脆性的;Ti3SiC2具有很强的抗体积变化的特性,Ti3AlC2的弹性模量的值相对较小,可以判断其原子间结合力较弱,而Ti3GeC2材料则有较高的弹性模量,说明其原子间结合力相对较强,材料的强度较大.
[1]Liu Y,Zhang J B,Li Y,Xiao X P,Chen H M 2015Mater.Rev.29 517(in Chinese)[刘耀,张建波,李勇,肖翔鹏,陈辉明2015材料导报29 517]
[2]Sezgin A,Aynur T,Yasemin O C 2016Solid State Sci.53 44
[3]Sitaram A,Ridwan S,Li Z O,Wai Y C 2015J.Eur.Ceram.Soc.35 3219
[4]Jiao Z Y,Ma S H,Wang T X 2015Solid State Sci.39 97
[5]Marcus H,Rolf G,Anneka V,Henry R,Peter S 2014Surf.Coat.Technol.257 286
[6]Navid A,Mina S H,Hamid R B,Naser E 2016Int.J.Refract.Met.Hard Mater.61 67
[7]Jeitschko W,Nowotny H,Die K Y 1967Monatsh.Chem.98 329
[8]Pietzka M A,Schuster J C 1994J.Phase Equilibria15 392
[9]Barsoum M W 2000Prog.Solid State Chem.28 201
[10]Benoit C,Ellen H,Nikhil K,Dominique V,Sylvain D 2005Powder Technol.157 92
[11]Payne M C,Clarke L J 1992Comput.Phys.Commun.72 14
[12]Segall M D,Lindan P J D,Probert M J 2002J.Phys.Condens.Matter14 2717
[13]Medvedeva N I,Freeman A J 2008Scr.Mater.58 671
[14]Yue L B,Xiao D H,Yue S,Chun C Z,Ming W L,Li P S 2010Solid State Sci.12 1220
[15]Jing R X,Chen X W,Teng F Y,Shu Y K,Jian M X,Yu G Y 2013Nucl.Instrum.Methods Phys.Res.Sect.B304 27
[16]Shou X C,Wen X F,Hai Q H,Gui Q Z,Zeng T L,Zi Z G 2011J.Solid State Chem.184 786
[17]Stojkovi M,Koteski V,Belovševi C,Čavor J 2008Phys.Rev.B77 193
[18]Xiao J K,Hua K,Chun B Z,Peter R 2015Chem.Phys.446 1
[19]Zhang H Z,Wang S Q 2007Acta Mater.55 4645
[20]Bai Y L,He X D,Sun Y,Zhu C C,Li M W,Shi L P 2010Solid State Sci.12 1220
[21]Sin’ko G V,Smirnov N A 2002J.Phys.Condens.Matter14 6989
[22]Neumann G S,Stixrude L 1999Phys.Rev.B60 791
[23]Xiao M Y,Hua H,Yu H Z,Ling Y,Pei D H 2014Comput.Mater.Sci.84 374
[24]Liu Y,Hu W C,Li D J,Zeng X Q,Xu C S,Yang X J 2012Intermetallics31 257
[25]Hu W C,Liu Y,Li D J,Zeng X Q,Xu C S 2013Physica B427 85
[26]Fan K M,Yang L,Sun Q Q,Dai Y Y,Peng S M,Long X G,Zhou X S,Zu X T 2013Acta Phys.Sin.62 116201(in Chinese)[范开敏,杨莉,孙庆强,代云雅,彭述明,龙兴贵,周晓松,祖小涛2013物理学报62 116201]
PACS:71.15.Mb,71.15.–m,71.15.Nc,71.20.–b DOI:10.7498/aps.66.057102
First principles study of electronic and elastic properties of Ti3AC2(A=Si,Sn,Al,Ge)phases∗
Hu Jie-Qiong Xie Ming†Chen Jia-Lin Liu Man-Men Chen Yong-TaiWang Song Wang Sai-BeiLi Ai-Kun
(State Key Laboratory of Advanced Technologies for Comprehensive Utilization of Platinum Metals,Kunming Institute of Precious Metals,Kunming 650106,China)
9 October 2016;revised manuscript
1 December 2016)
A first-principles plane-wave pseudo potential method based on the density functional theory is used to investigate the phase structures,energies,electronic structures and elastic properties of Ti3AC2(A=Si,Sn,Al,Ge)phases.In this paper,Ti3AC2(A=Si,Sn,Al,Ge)crystal structures are first optimized,then the band structures,total and part density of states,charge density distributions and elastic properties of these compounds are analyzed,and the cohesive energies and formation energy of these phases are also calculated.The results show that the Ti3GeC2is more stable than other compounds,the formation energy of Ti3AlC2is the lowest in these compounds,which indicates that Ti3AlC2is easier to generate;Ti3AC2(A=Si,Sn,Al,Ge)each have a higher density of states at Fermi level,which shows the strong metallicity,meanwhile,the electrical conductivity of each phase is anisotropic.The DOS at the Fermi energy is mainly from the Ti-d electrons,which should be involved in the conduction properties although d electrons are considered to be inefficient conductors.The lowest valence bands are formed by the C-s states with a small mixture of Ti-p+d,andA-s+p states.The electrical properties are mainly decided by the p-d hybridizations between 3d electrons in Ti and the p electrons inA(A=Si,Sn,Al,Ge)and 2p electrons in C,and the strong hybridization of p-d states make the materials have stable structures.It should be noted that the calculated bond length of Ti-Ge is shorter than those of Ti—A(A=Si,Sn,Al)bonds.This implies that the Ti—Ge bond is stronger than Ti—A(A=Si,Sn,Al)bonds.Furthermore,the Fermi level of Ti3GeC2is relatively low,which also indicates the relatively high stability of Ti3GeC2.The charge density provides a measure of the strength of the ionic bond,so that Ti3GeC2and Ti3SiC2have stronger ionic bonds than Ti3SnC2and Ti3AlC2.The strongM—Abonds in Ti3GeC2lead toadecreasing andclattice parameter value increasing.The spherical shape ofXrepresents more like an ionic bond.Thez-directional localized shapes ofAeach is more like a covalent bond.The covalent bonds ofAelements each are localized along thezdirection so that they affect mostly theclattice parameter;the calculated elastic properties of Ti3AC2(A=Si,Sn,Al,Ge)phases show that the atomic binding force of Ti3AlC2is weaker than those of other three phases,while the atomic binding force of Ti3GeC2is relatively strong,which makes the strength of Ti3GeC2quite high.
first-principles,MAXphases,electronic structures,elastic properties
PACS:71.15.Mb,71.15.–m,71.15.Nc,71.20.–b
10.7498/aps.66.057102
∗国家自然科学基金(批准号:U1302272,51267007,51461023)、云南省院所技术开发专项(批准号:2013DC016)和稀贵金属材料协同创新基金项目(批准号:2014XT02)资助的课题.
†通信作者.E-mail:joanr8210@163.com
*Project supported by the National Natural Science Foundation of China(Grant Nos.U1302272,51267007,51461023),the Institutes Special Technology Development Project of Yunnan,China(Grant No.2013DC016),and the Fund of the Collaborative Innovation Center of Rare and Precious Metals Advanced Materials,China(Grant No.2014XT02).
†Corresponding author.E-mail:joanr8210@163.com
