心脏肌球蛋白结合蛋白C基因c.G772A突变与家族性肥厚型心肌病
2017-07-31邢晓博刘福颂王芳宋雷赵雯娜刘杰张克传朱玉召刘歆袁帅孙璐尚兴福李荣梁琰李晓樊光红张长青
邢晓博,刘福颂,王芳,宋雷,赵雯娜,刘杰,张克传,朱玉召,刘歆,袁帅,孙璐,尚兴福,李荣,梁琰,李晓,樊光红,张长青
心脏肌球蛋白结合蛋白C基因c.G772A突变与家族性肥厚型心肌病
邢晓博,刘福颂*,王芳,宋雷,赵雯娜,刘杰,张克传,朱玉召,刘歆,袁帅,孙璐,尚兴福,李荣,梁琰,李晓,樊光红,张长青
目的:研究中国人肥厚型心肌病(HCM) 患者致病基因突变位点,并分析基因型与临床表型的关系。
方法:在HCM家系中利用靶向外显子捕获测序的方法对HCM先证者的30个与遗传性心肌病相关的基因进行全外显子扩增和高通量测序,进一步通过Sanger测序法在家系内及200例健康志愿者中进行验证。家系调查资料包括临床表现、体格检查、心电图及超声心动图。
结果:该家系6例有血缘关系的研究对象中3例携带心脏型肌球蛋白结合蛋白C基因(MYBPC3) c.G772A杂合突变,该突变位点位于MYBPC3的258位的谷氨酸(E)变为赖氨酸(K)。其余家系成员未发现此突变。200例健康志愿者中未见异常。先证者及其女儿发病年龄晚且均伴有心悸、胸闷的症状,超声心动图示室间隔基底段增厚(16~18 mm)。先证者目前伴有阵发性室性心动过速恶性心律失常及心力衰竭,左心室流出道最大压差为56 mmHg(1 mmHg=0.133 kPa),属于猝死高危人群。
结论:全面基因检测有利于临床危险分层及早诊治。MYBPC3 的剪切位点突变c.G772A可能是该HCM家系的致病突变。
心肌病,肥厚性;基因突变
(Chinese Circulation Journal, 2017,32:680.)
肥厚型心肌病(HCM)是最常见的常染色体显性遗传性心肌病,以编码肌小节蛋白基因突变引起的收缩力产生缺陷为主要病因,涉及至少20个基因和1 400多个突变位点,发病率约为1:500[1,2]。心脏型肌球蛋白结合蛋白C基因(MYBPC3)是HCM的主要致病基因,约占15%~25%。 MYBPC3突变的HCM患者心肌肥厚出现晚、症状轻,不利于临床诊断,由此可能延误病情,而基因诊断是有用的手段之一[3-5]。HCM人群发病率高,有潜在凶险表型,本研究通过高通量靶向捕获测序在一家系发现MYBPC3 c.G772A,分析突变基因型与临床表型的关系,为HCM危险分层及治疗评估提供了依据。
1 资料与方法
1.1研究对象及临床资料收集
选择中国汉族家族性HCM一家系(图1),临床资料包括体检、心电图、超声心动图等结果。HCM诊断标准符合2011年美国心脏病学院基金会(ACCF)/美国心脏协会(AHA)HCM诊断和治疗指南的诊断标准,即临床上不能解释的、无心室腔扩张的左心室肥厚(超声心动图示左心室壁厚度≥15 mm),且无其他导致心室肥厚的心脏疾病或系统性疾病[6]。同时满足2014年欧洲心脏病学会HCM诊断与治疗指南的诊断标准[7]。选择200例健康志愿者为正常对照,所有研究对象已签署知情同意书。
1.2方法
提取基因组DNA: 采集HCM患者及正常对照者外周静脉血5 ml,置于含有乙二胺四乙酸(EDTA)的抗凝管中备用,采用吸附法DNA提取试剂盒(QIAamp DNA Blood Mini Kit , QIAGEN公司,德国)提取血液样本中的DNA,经过琼脂糖电泳鉴定样本DNA质量合格后置-20℃冰箱中保存。
基因分析:用3.0 μg DNA作为起始,根据Agilent’s SureSelect XT target enrichment system的实验流程来制备文库,再使用设计好的探针(诺心安panel Bestnovo公司,中国)对目标区域完成捕获,利用靶向外显子捕获测序技术对30个与遗传性心肌病相关的基因按照Illumina-SolexaHiSeq2000程序装置进行第2代测序( Next generation sequencing NGS),测序仪器为MiSeq System(Illumina公司,美国)。运用http://www.ncbi.nlm.nih.gov/blast/bl2seq程序来分析测序结果,明确突变位点,进一步通过一代测序(Sanger测序法)在家系主要成员及200例健康志愿者中进行验证。
2 结果
2.1测序结果(图1、2)
在人类基因组变异协会(HGVS)标准基础上与参考基因组NCBI Genome(http://www.ncbi. nlm.nih.gov/genome)比对,并综合考虑人群频率数 据 库 Exome Aggregation Consortium(http://exac. broadinstitute.org/)/1000 Genomes Project和致病性数据 库 ClinVar(http://www.ncbi.nlm.nih.gov/clinvar)/ OMIM(http://www.omim.org),由我们的遗传咨询团队评估得出结果。3例患者携带MYBPC3 c.G772A,使258位的谷氨酸(E)变为赖氨酸(K)。该突变点为高度保守序列。其余家系成员不携带突变基因,200例正常对照者中未见异常。
2.2家系成员临床表现(表1)
该家系中先证者(I-2)为75岁女性患者,因“反复发作心悸、胸闷10年”就诊。24 h动态心电图示阵发性室性心动过速(室速)。超声心动图示收缩期前向运动(SAM)征阳性,最大室壁厚度18.3 mm,左心室短轴缩短率(FS)35%,左心室舒张功能减退。冠状动脉计算机断层血管摄影术示符合冠状动脉硬化表现,右冠状动脉走行区见点状钙化,相应管腔未见明显变窄,左冠状动脉无异常。先证者女儿(II-2),50岁,因“反复发作心悸、胸闷5年”就诊。 超声心动图示SAM征阳性,最大室壁厚度18.0 mm,FS 36%,左心室舒张功能减退。冠状动脉计算机断层血管摄影术示符合冠状动脉硬化表现,左冠状动脉回旋支近端似斑块形成并狭窄,远端闭塞。
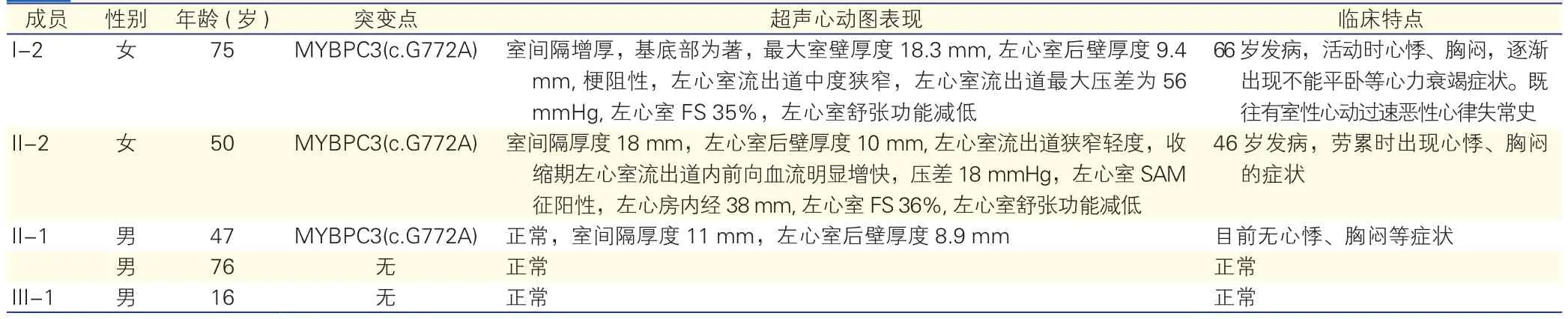
表1 携带MYBPC3基因c.G772A突变的HCM家系成员临床资料
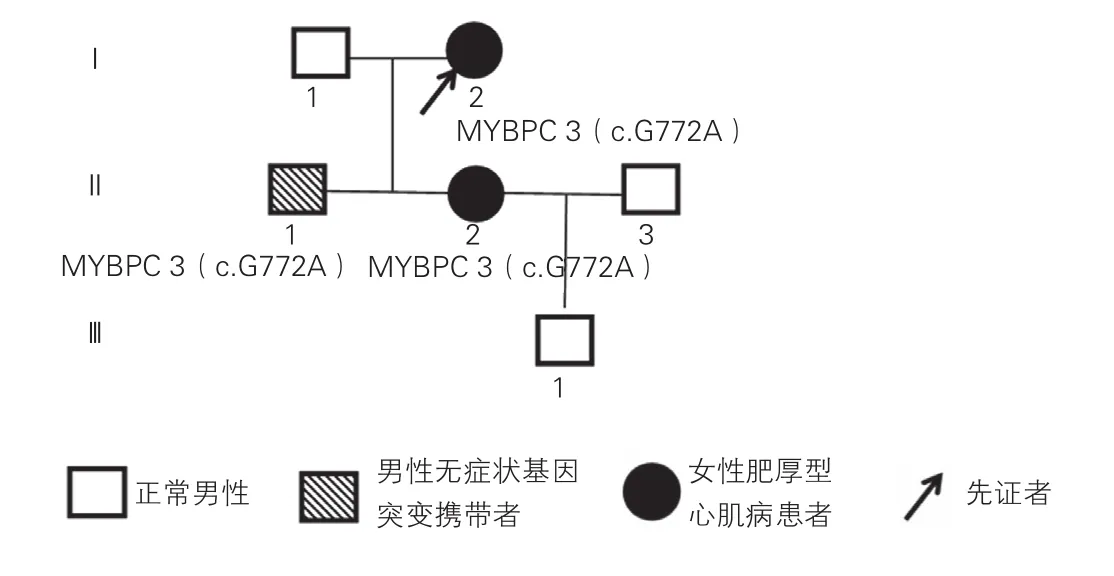
图1 一家族性肥厚型心肌病家系系谱图
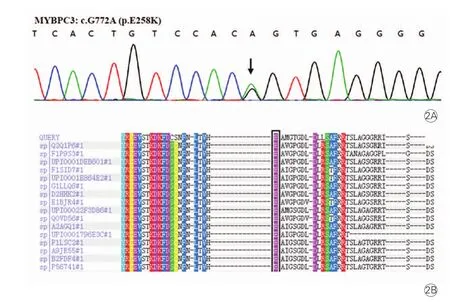
图2 测序结果图
3 讨论
MYBPC3 编码心肌肌球蛋白结合蛋白 C,该蛋白仅在心肌中表达,其C1 IgI区C2 IgI区连接部称“基序(motif)”区,系9个氨基酸残基组成的环状结构,是环磷酸腺苷依赖性蛋白激酶A及钙调蛋白依赖性蛋白激酶进行底物可逆性磷酸化的关键位点,其磷酸化可调节心肌收缩[8]。该HCM家系携带MYBPC3 c.G772A,是C1IgI区6外显子最后一个核苷酸剪切位点突变,导致258位的谷氨酸(E)变为赖氨酸(K),为罕见变异,其在千人基因组数据库、ESP6500 数据库均无人群频率报道,ExAC 数据库的人群频率为 0.00003903。该变异曾被多个研究表明是HCM的致病变异,MYBPC3 c.G772A携带者部分为家系筛查时发现存在心肌肥厚, 有些携带者则并无明显心肌肥厚等异常,外显率不完全,并且发病与年龄有关,随着年龄增加而外显率增加[9-11]。Marston等[12]研究发现携带该变异的患者心肌组织中的心肌肌球蛋白结合蛋白 C 含量下降了24%,由于单倍剂量不足而致病。
本家系资料表明,该位点突变在这一家系中的特点为临床发病晚(先证者66岁发病,其女儿46岁发病,其儿子在47岁还未发病),与既往研究“MYBPC3基因突变携带者发病较晚(>40岁)且中老年发病多见”一致[13-14]。值得注意的是,虽然HCM自然病程存在明显的异质性,大约25%的患者临床无症状或仅有轻微的症状,并可以享有正常的寿命(75岁以上),但最终大多数患者会发生心力衰竭、心房颤动导致卒中甚至猝死[15-16]。该研究中先证者晚年发病开始疑诊为冠心病,提示基因突变筛查是HCM诊断的重要补充。2014年欧洲心脏病学会HCM诊断和管理指南指出,HCM是临床和基因表现复杂的疾病,基因检测有利于HCM的诊断、危险分层及心脏性猝死的早期预警,建议强化基因检测和遗传咨询,在进行家系的临床筛查时,尤其年轻携带者,建议制定长期的随访计划[7]。
[1] van der Velden J, Ho CY, Tardiff JC, et al. Research priorities in sarcomeric cardiomyopathies . Cardiovasc Res, 2015, 105: 449-456.
[2] Bottillo I , D'Angelantonio D , Caputo V , et al. Molecular analysis of sarcomeric and non-sarcomeric genes in patients with hypertrophic cardiomyopathy. Gene, 2016 , 577: 227-235.
[3] Manrai AK , Funke BH , Rehm HL , et al. Genetic misdiagnoses and the potential for health disparities. N Engl J Med, 2016, 375: 655-665. [4] 陈石, 乔树宾. 肥厚型心肌病左心室舒张功能障碍研究进展. 中国循环杂志, 2013, 28 : 316-318.
[5] 安硕研, 樊朝美, 赵世华, 等. 左心室中部肥厚型梗阻性心肌病的临床特点及预后. 中国循环杂志, 2015, 30 : 1053-1057.
[6] Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol, 2011, 58: e212-260.
[7] Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC) . Eur Heart J, 2014, 35: 2733-2779.
[8] Flashman E, Redwood C, Moolman-Smook J, et al. Cardiac myosin binding protein C: its role in physiology and disease. Circ Res, 2004, 94: 1279-1289.
[9] Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med, 1998, 338: 1248-1257.
[10] Song L, Zou Y, Wang J, et al. Mutations profile in Chinese patients with hypertrophic cardiomyopathy. Clin Chim Acta, 2005, 351: 209-216.
[11] Page SP, Kounas S, Syrris P, et al. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet, 2012, 5: 156-166.
[12] Marston S, Copeland O, Jacques A, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res, 2009, 105: 219-222.
[13] 李静, 刘丽文, 纳丽莎, 等. 心脏肌球蛋白结合蛋白C基因P1208fs突变与家族性肥厚型心肌病的关系 . 中华心血管病杂志, 2016, 44: 321-326.
[14] Page SP, Kounas S, Syrris P, et al. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet, 2012, 5: 156-166.
[15] Rubattu S, Bozzao C, Pennacchini E, et al. A next-generation sequencing approach to identify gene mutations in early- and lateonset hypertrophic cardiomyopathy patients of an Italian cohort. Int J Mol Sci, 2016, 17: E1239.
[16] McTaggart DR, Ogden KJ, Marathe JA. A long term follow-up study of carriers of hypertrophic cardiomyopathy mutations. Heart Lung Circ, 2017, 26: 18-24.
Relationship Between Cardiac Myosin-binding Protein c.G772A Gene Mutation and Familial Hypertrophic Cardiomyopathy
XING Xiao-bo, LIU Fu-song, WANG Fang, SONG Lei, ZHAO Wen-na, LIU Jie, ZHANG Ke-chuan, ZHU Yu-zhao, LIU Xin, YUAN Shuai, SUN Lu, SHANG Xing-fu, LI Rong, LIANG Yan, LI Xiao, FAN Guang-hong, ZHANG Chang-qing.
Department of Cardiology, The Third People’s Hospital of Qingdao City, Qingdao (266004), Shandong, China
WANG Fang, Email: wf8522@126.com
Objective: To investigate the mutation site of pathogenic gene in patients with hypertrophic cardiomyopathy (HCM) and to analyze the relationship between the genotype and clinical phenotype.
Methods: Targeted exon capture sequencing was conducted in a HCM proband for 30 coding exons related HCM gene by all exon amplification and high-throughput sequencing. Furthermore, Sanger sequencing was performed in other family member and in 200 healthy volunteers for verification. The familial investigation included in clinical presentation, physical examination, electrocardiogram and echocardiography.
Results: There were 3/6 blood relatives carrying cardiac myosin-binding protein gene MyBPC3 G772A heterozygous mutation, the mutation site was at 258 amino acid of MyBPC3 as glutamic acid (Glu) was substitute to lysine (Lys), such mutation was not found in rest of family member and not in healthy volunteers. The onset of proband and her daughter was rather late, they had palpitation and chest tightness; echocardiography showed interventricular septum basal segment thickening (16-18) mm. Proband was complicating paroxysmal ventricular tachycardia, malignantarrhythmia and heart failure, the maximum pressure gradient of left ventricular outflow was 56 mmHg, which with the high risk for sudden death.
Conclusion: Comprehensive gene test has been helpful for clinical stratification, early diagnosis and treatment. MYBPC3 site mutation c.G772A might be the pathogenic mutation in that specific HCM family.
Cardiomyopathy, hypertrophic; Gene; Mutation
2016-08-20)
(编辑:常文静)
青岛市科研计划(2015-WJZD061)
266041 山东省,青岛市第三人民医院 心内科(邢晓博 、刘福颂、王芳、赵雯娜、刘杰、刘歆、袁帅、孙璐、尚兴福、李荣、梁琰、李晓、樊光红、张长青),检验科(张克传),特检科(朱玉召);中国医学科学院 北京协和医学院 国家心血管病中心 阜外医院 高血压诊治中心(宋雷)
邢晓博 主任医师 学士 主要研究方向冠心病、高血压和肥厚型心肌病 Email:13668884192@126.com 通讯作者:王芳 Email: wf8522@126.com*刘福颂为共同第一作者
R596
A
1000-3614(2017)07-0680-04
10.3969/j.issn.1000-3614.2017.07.014
