EMRE在小鼠心肌缺血损伤中的作用及其相关机制的研究
2017-07-31刘峰舟荆哲刘明莉张恩尉宁江华牟佼
刘峰舟,荆哲,刘明莉,张恩尉,宁江华,牟佼
EMRE在小鼠心肌缺血损伤中的作用及其相关机制的研究
刘峰舟,荆哲,刘明莉,张恩尉,宁江华,牟佼
目的:研究调节线粒体钙单向转运体活性所必须的蛋白(essential MCU regulator,EMRE)在小鼠心肌缺血损伤中发挥的作用及其相关机制。
方法:利用心肌点注射EMRE腺病毒(Ad-EMRE)上调心肌组织的EMRE分子表达水平,对照小鼠心肌点注射增强绿色荧光蛋白腺病毒(Ad-eGEP),48 h后,采用结扎小鼠左冠状动脉前降支方法建立心肌梗死(MI)模型。实验设置三组:假手术组(给予Ad-eGFP);心肌梗死对照组(给予Ad-eGFP);心肌梗死处理组(给予Ad-EMRE)。三周后,利用小动物超声系统测定小鼠心脏功能;Western blot及免疫组化检测EMRE分子的表达;麦胚凝集素(WGA)染色检测心肌细胞肥大程度;马松(MASSON)染色检测心肌组织胶原含量;原位末端标记法(TUNEL)检测心肌组织的细胞凋亡;Western blot检测心肌组织半胱氨酸天冬氨酸蛋白酶-3(Caspase-3)和半胱氨酸天冬氨酸蛋白酶-9(Caspase-9)的表达;MitoSOX荧光探针检测线粒体ROS水平。
结果:与心肌梗死对照组相比,心肌梗死处理组的心功能明显变差,心肌肥厚明显加重,心肌组织胶原纤维含量更高。另外,心肌梗死处理组的心肌细胞凋亡明显增多,Caspase-3和Caspase-9表达明显升高,线粒体活性氧簇产生也明显增多。
结论:EMRE过表达可增加线粒体ROS生成,诱导心肌细胞凋亡,加重心肌缺血损伤。
线粒体转运蛋白质类;心肌缺血
(Chinese Circulation Journal,2017,32:701.)
心肌缺血损伤是心血管系统疾病的发病机制之一[1],然而心肌缺血损伤的机制尚未阐明清楚。目前的研究表明,线粒体钙超载,线粒体损伤,细胞凋亡和坏死等在心肌缺血损伤的发生、发展过程中发挥着重要的作用[2,3]。调节线粒体钙单向转运体活性所必须的蛋白(essential MCU regulator,EMRE)位于线粒体内膜,单次跨膜蛋白,维持线粒体钙单向转运体( mitochondrial calcium uniporter,MCU)的正常功能,参与线粒体钙稳态的调控[4,5]。目前,EMRE在心肌缺血损伤中的作用鲜有报道,为了明确EMRE在心肌缺血损伤中的作用,本研究拟在小鼠心肌点注射EMRE腺病毒(Ad-EMRE)上调EMRE表达的基础上,建立心肌梗死模型,以此来探讨EMRE在心肌缺血损伤中的作用及其相关机制,期望为临床心肌缺血损伤的防治提供新的靶点和方向。
1 材料与方法
1.1实验材料
8周龄雄性C57BL/6小鼠30只购自第四军医大学动物中心,微量注射器购自瑞士汉密尔顿公司,Ad-EMRE和Ad-增强绿色荧光蛋白(eGFP)均为本实验室已有,EMRE抗体购自美国Santa cruz公司,半胱氨酸天冬氨酸蛋白酶-3(Caspase-3)、半胱氨酸天冬氨酸蛋白酶-9(Caspase-9)和β-肌动蛋白(β-actin)抗体及辣根过氧化物酶(HRP)标记的二抗购自武汉三鹰公司,电化学发光(electrochemiluminescence,ECL)液购自美国Advansta公司,聚偏二氟乙烯膜(polyvinylidene fluoride,PVDF)购自美国Invitrogen公司,蛋白定量、细胞总蛋白提取试剂盒,购自上海碧云天公司,麦胚凝集素( wheat germ agglutinin,WGA)购自美国Sigma公司,原位末端标记法(TUNEL)凋亡检测试剂盒购自瑞士罗氏公司,4,6-二脒基-2-苯基吲哚(DAPI)核标记染料购自美国Invitrogen公司,MitoSOX Red荧光探针购自美国Invitrogen公司,PBS缓冲液(pH=7.4;主要成分:NaCl 137mmol/L,KCl 2.7 mmol/L,Na2HPO410 mmol/L, KH2PO42 mmol/L)实验室配制。
1.2方法
实验分组及EMRE表达上调:将30只8周龄雄性C57BL/6小鼠随机分为三组(每组10只):假手术组(给予Ad-eGFP);心肌梗死对照组(给予AdeGFP);心肌梗死处理组(给予Ad-EMRE)。经小鼠左胸第4~5肋间暴露心脏,利用微量注射器在小鼠左心室心肌壁内分别注射Ad-EMRE和Ad-eGFP(30 μl),然后将心脏置回并排空胸腔气体,采用荷包法缝合伤口。48 h后,每组随机选取3只,提取心肌组织蛋白,蛋白免疫印迹(Western blot)法检测EMRE表达水平。
心肌梗死小鼠模型建立:小鼠心肌点注射Ad-EMRE 48 h后,在左胸同一位置暴露心脏,通过结扎左冠状动脉(冠脉)前降支建立心肌梗死模型,术后心电图显示心肌出现缺血,即确认心梗模型成功建立。
心功能检测:利用异氟烷麻醉小鼠,待其状态稳定后,采用加拿大Visual Sonics公司所产的Vevo 2100高分辨率小动物超声成像系统检测小鼠心功能变化,利用Vevo 2100系统分析软件进行数据的处理分析,测量小鼠左心室射血分数(LVEF)和左心室短轴缩短率(LVFS)。
心肌组织学分析:取小鼠心脏,用预冷的PBS缓冲液冲洗心脏,取冠脉结扎部位以下心肌组织,用10%的福尔马林溶液固定48 h后,进行挂机,包埋,制作石蜡切片。冰冻切片则无需固定,用包埋剂处理新鲜心肌组织后,进行制片。麦胚凝集素(wheat germ agglutinin,WGA)对小鼠心脏组织的冰冻切片进行染色检测心肌细胞肥大情况,WGA可以特异性的标记心肌细胞膜,呈现红色荧光,显示心肌细胞横截面的轮廓,共聚焦显微镜观察拍照,利用Image-Pro Plus 6软件进行数据分析计算心肌细胞的平均横截面积[6,7]。马松(MASSON)染色检测心肌组织纤维化水平,光学显微镜下,每张切片随机选取六个视野进行观察拍照,Image-Pro Plus 6软件分析心肌组织胶原纤维容积分数,胶原纤维容积分数=胶原纤维面积/视野总面积。
TUNEL荧光标记法检测心肌细胞凋亡:按照试剂盒说明处理标本进行检测,DAPI可以穿透细胞膜与细胞核中的双链DNA结合标记为蓝色荧光,TUNEL可以特异性的将凋亡细胞中损伤断裂的基因组DNA标记为绿色荧光,当心肌细胞核被同时标记为蓝色和绿色荧光时提示该心肌细胞发生了凋亡。
心肌组织蛋白提取及Western blot 法检测EMRE分子的表达:取左前降支结扎部位以下的心肌组织,预冷PBS缓冲液洗两遍,匀浆器匀浆,再次PBS缓冲液清洗一遍后,离心去上清,加入裂解液(含蛋白酶抑制剂和磷酸酶抑制剂),冰上裂解半小时,余步骤同细胞蛋白提取一致。随后进行聚丙烯酰胺凝胶电泳,EMRE 抗体稀释比例为1∶500,Caspase-3 抗体稀释比例为 1∶1 000,Caspase-9 抗体稀释比例为 1∶800,β-actin 抗体稀释比例为 1∶3 000,采用ECL化学发光试剂盒进行检测。
心肌细胞线粒体活性氧簇(MitoROS)检 测:利 用 荧 光 探 针MitoSOX Red(Invitrogen)对小鼠心肌组织的石蜡切片进行染色,检测心肌细胞线粒体的活性氧(ROS)水平。MitoSOX Red是一种特异性靶向线粒体的新型荧光染料。MitoSOX Red被超氧化物氧化,氧化之后可以与核酸结合发出红色的荧光。因此,我们可以通过检测红色荧光的强弱来反映线粒体ROS的水平。红色荧光越强,提示MitoROS水平越高,MitoSOX Red(5μmol/L)室温避光孵育15min,PBS缓冲液洗片后,共聚焦显微镜拍照,并利用Image-Pro Plus 6软件进行荧光强度的分析和处理[8]。
统计学方法:采用SPSS16.0统计学软件进行数据处理。数据均以均数 ± 标准差( ±s)表示,组间比较采用单因素方差分析( one-way ANOVA),P<0.05为差异具有统计学意义。
2 结果
2.1上调EMRE对心肌梗死模型小鼠心脏功能的影响(图1)
在C57小鼠心肌点注射Ad-EMRE腺病毒48h后,Western blot检测心肌组织EMRE的表达水平。结果显示,心肌梗死处理组小鼠心肌组织中的EMRE表达水平明显高于心肌梗死对照组的小鼠(P<0.01)。心肌梗死三周后,小动物超声结果显示,与心肌梗死对照组相比,心肌梗死处理组的左心室射血分数明显较弱(P<0.01),短轴缩短率较低(P<0.01),心脏功能明显变差。
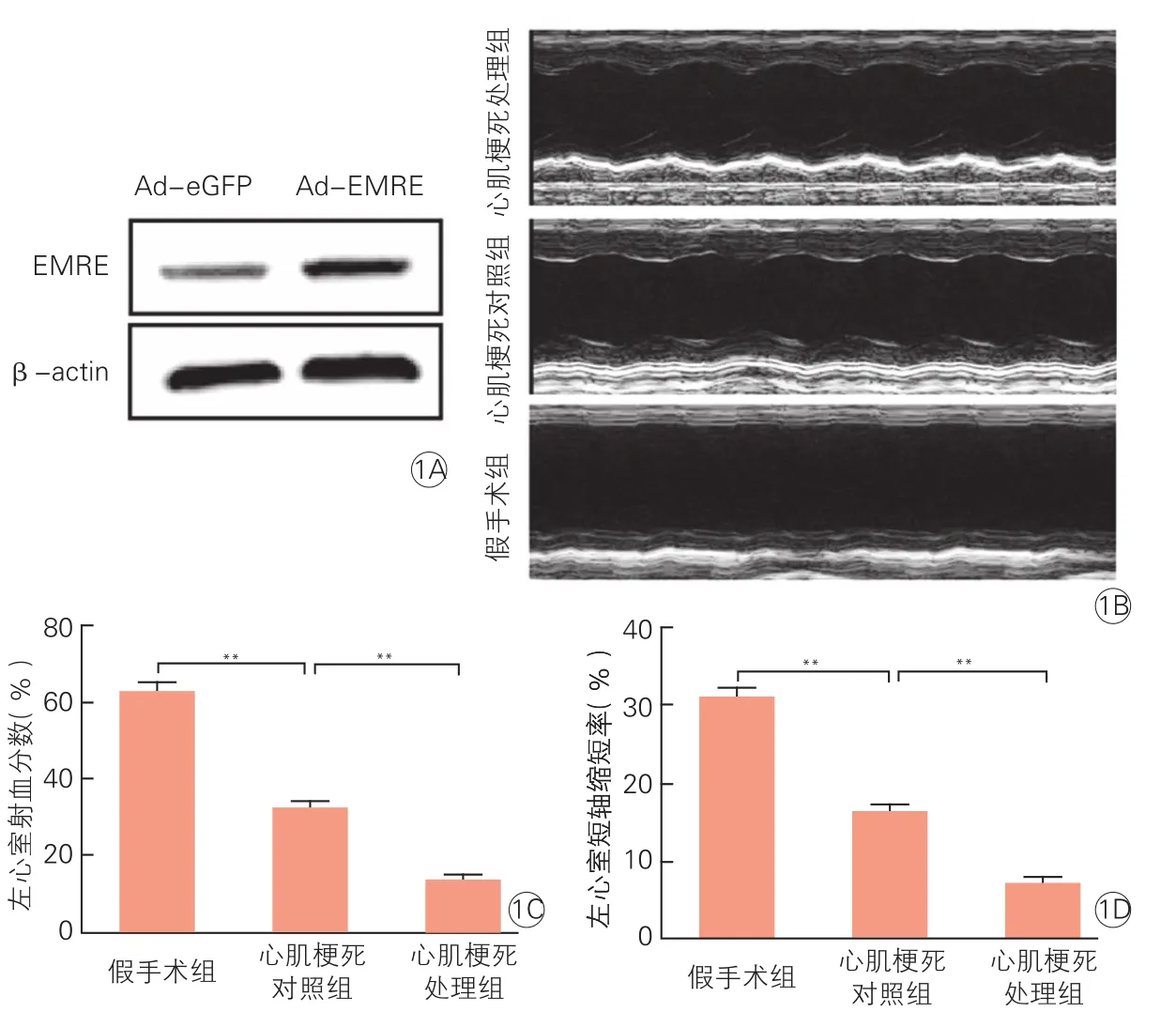
图1 过表达EMRE加重心肌损伤
2.2EMRE过表达促进心肌梗死模型小鼠心肌细胞肥大,加重心肌纤维化水平(图2)
小鼠心肌缺血三周后,心肌组织免疫组化检测EMRE表达水平;与其他两组比较,EMRE在心肌梗死处理组表达水平最高(P<0.01);Masson染色结果显示,心肌细胞呈现红色,胶原纤维呈现蓝色,可以看出心肌梗死处理组的心肌组织胶原含量较心肌梗死对照组相比也明显增多(P<0.01),纤维化水平升高,WGA染色结果显示,与心肌梗死对照组比,心肌梗死处理组的心肌细胞平均横截面积明显增大,提示该组的心肌细胞肥大更加明显(P<0.01)。
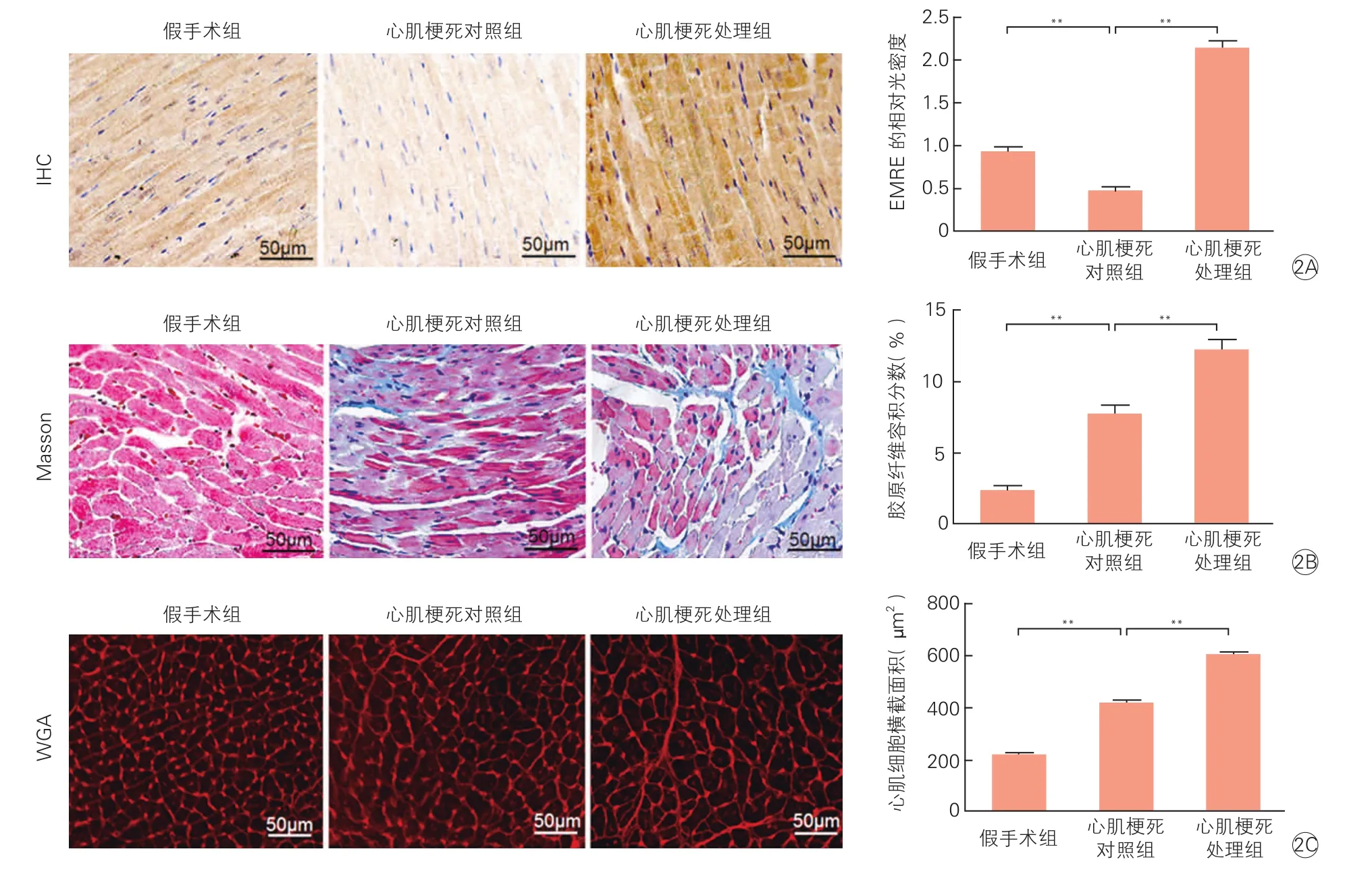
图2 过表达EMRE加重心肌细胞肥大
2.3EMRE过表达对心肌细胞凋亡的影响(图3)
TUNEL法检测心肌组织细胞凋亡水平,DAPI染料与细胞核中的双链DNA结合将心肌细胞核被标记为蓝色,当心肌细胞发生凋亡时,损伤断裂的核基因组DNA可被TUNEL标记为绿色荧光,心肌梗死处理组的心肌细胞凋亡水平明显高于心肌梗死对照组和假手术组,提示EMRE过表达可以促进心肌细胞凋亡; Western blot检测结果显示,心肌梗死处理组的Caspase-3表达水平明显高于心肌梗死对照组(P<0.01),和TUNEL检测结果一致,Caspase-9的表达水平也明显升高(P<0.01),提示EMRE过表达促进心肌细胞凋亡是通过线粒体途径。

图3 过表达EMRE促进心肌细胞凋亡
2.4EMRE过表达促进缺血导致的心肌细胞线粒体ROS生成(图4)
利用MitoSOX Red染料对心肌组织的石蜡切片进行染色检测线粒体ROS水平,心肌梗死处理组的红色荧光强度明显高于心肌梗死对照组,提示该组心肌细胞线粒体ROS生成明显增加(P<0.01)。进一步说明EMRE过表达导致心肌细胞线粒体ROS水平增高,从而激活了线粒体途径的细胞凋亡。
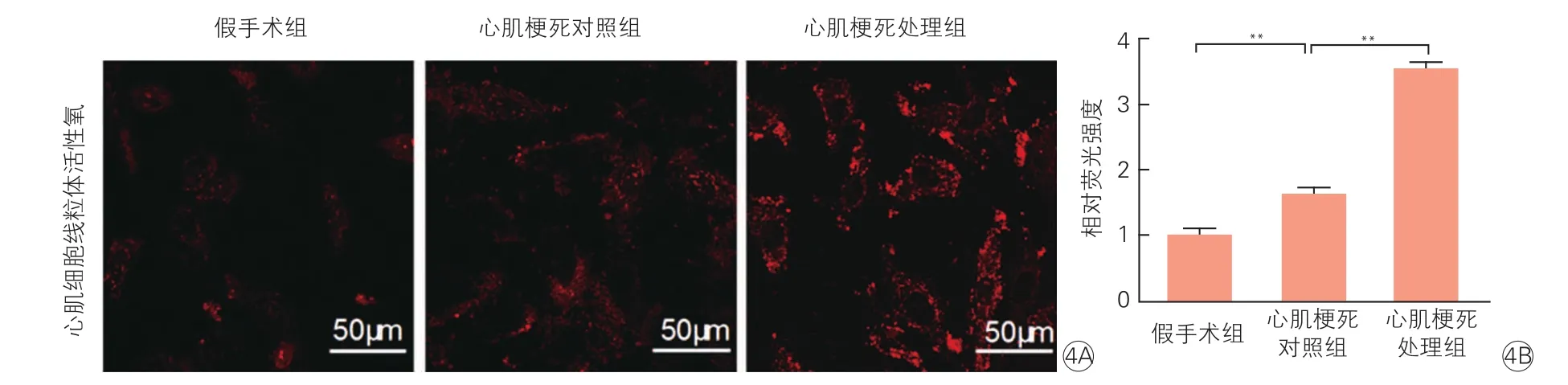
图4 心肌组织石蜡切片染色显示结果过表达EMRE促进心肌细胞线粒体活性氧的生成
3 讨论
缺血性心脏病已成为致死和致残的主要原因[9]。因此,深入研究心肌缺血损伤的干预靶点和治疗措施是很有必要的。线粒体钙信号参与调节细胞的多种生命活动,目前研究认为线粒体钙稳态与线粒体功能障碍,细胞死亡等密切相关[10,11]。线粒体功能紊乱与心肌缺血的发生及进展密切相关,是心肌缺血损伤的重要原因之一[12]。
EMRE在线粒体钙稳态调控中发挥重要作用,维持线粒体功能的正常发挥。我们前期研究发现在高糖高脂处理的H9C2细胞中,下调EMRE可以减少线粒体ROS的生成,抑制心肌细胞的凋亡[13]。但是在心肌缺血损伤中,EMRE发挥的作用及其相关机制尚不清楚,因此,我们推测EMRE通过影响线粒体功能参与了心肌缺血损伤的病理过程。
我们采用心肌点注射腺病毒方式,在小鼠心脏过表达EMRE分子,48 h后,每组随机选择三只小鼠,并提取心肌组织蛋白,采用Western blot方法检测确认EMRE分子的过表达效果。在此基础上,我们选择结扎左前降支方式建立缺血模型,研究上调EMRE分子对心肌缺血损伤的影响。小动物超声检测小鼠心脏功能发现,过表达EMRE分子后,心肌梗死模型小鼠的左心室射血分数明显低于对照组及假手术组,通过WGA染色和MASSON染色,我们观察到心肌梗死处理组的小鼠的心肌细胞肥大及心肌组织的纤维化水平与心肌梗死对照组和假手术组相比有明显的加重。进一步对心肌组织进行TUNEL检测发现EMRE过表达后的心肌细胞凋亡水平也明显高于心肌梗死对照组。结果表明,上调EMRE可以明显加重小鼠心肌缺血损伤,并且可能是通过增加心肌细胞的凋亡来参与心肌缺血损伤的病理过程。为了进一步研究相关机制,我们利用Western blot检测了凋亡相关分子Caspase-3和Caspase-9 的表达水平,发现其在心肌梗死处理组的表达水平显著高于其他组,进一步说明了上调EMRE可以引起内源性的心肌细胞凋亡。随后,我们利用MitoSOX Red荧光探针对线粒体的ROS水平进行了检测,发现EMRE的上调可以明显增加心肌细胞线粒体ROS的生成。目前的研究表明,线粒体钙超载在心肌缺血损伤病程进展中发挥着重要作用,抑制线粒体钙超载会减轻心肌缺血导致的损伤[3,14],EMRE在调控线粒体钙稳态中发挥着重要了作用,EMRE 的高表达致使线粒体钙单向转运体活性增高,进一步导致线粒体中钙离子水平升高而使线粒体钙超载,而线粒体钙超载又是导致线粒体ROS生成增加的一个重要原因[3]。我们认为,在心肌缺血条件下,EMRE高表达进一步加重了线粒体的钙超载,导致心肌梗死处理组的线粒体ROS水平明显高于心肌梗死对照组,进而加重心肌缺血损伤。
总之,EMRE是近年来新发现的线粒体钙摄取相关蛋白,在线粒体钙稳态调控中发挥着重要的作用[4],我们通过上调小鼠心肌组织中的EMRE表达水平发现,EMRE高表达可以通过诱导线粒体ROS水平升高,促进心肌细胞凋亡,从而加重心肌缺血损伤。本研究的局限性在于,我们只是观察了EMRE高表达水平下心肌损伤的变化过程,并没有对EMRE下调或者缺失条件下的心肌缺血损伤做进一步的研究,我们将在后续研究中,利用敲除动物模型对EMRE分子在小鼠心肌缺血损伤的作用机制进行更深层次的研究,期望对心肌缺血损伤的防治提供新的治疗靶点和方向。
[1] Dominguez-Rodriguez A, Abreu-Gonzalez P, Reiter RJ. Cardioprotection and pharmacological therapies in acute myocardial infarction: challenges in the current era. World J Cardiol, 2014, 6: 100-106.
[2] Aldakkak M, Stowe DF, Chen Q, et al. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+overload and ROS release. Cardiovasc Res, 2008, 77: 406-415.
[3] Santulli G, Xie W, Reiken SR, et al. Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci U S A, 2015, 112: 11389-11394.
[4] Sancak Y, Markhard AL, Kitami T, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science, 2013, 342: 1379-1382.
[5] Criddle DN,Tepikin AV. Intramitochondrial Ca (2+) sensing by EMRE: the matrix outlook on stimulus-metabolism coupling. Mol Cell, 2016, 61: 646-647.
[6] Zhou H, Yang HX, Yuan Y, et al. Paeoniflorin attenuates pressure overload-induced cardiac remodeling via inhibition of TGF beta/ Smads and NF-kappaB pathways. J Mol Histol, 2013, 44: 357-367.
[7] Luo Y, Zhao X, Zhou X, et al. Short-term intermittent administration of CXCR4 antagonist AMD3100 facilitates myocardial repair in experimental myocardial infarction. Acta Biochim Biophys Sin(Shanghai), 2013, 45: 561-569.
[8] Ramirez E, Klett-Mingo M, Ares-Carrasco S, et al.Eplerenone attenuated cardiac steatosis, apoptosis and diastolic dysfunction in experimental type-II diabetes. Cardiovasc Diabetol, 2013, 12: 172.
[9] Weir RA, McMurray JJ,Velazquez EJ. Epidemiology of heart failure and left ventricular systolic dysfunction after acute myocardial infarction: prevalence, clinical characteristics, and prognostic importance. Am J Cardiol, 2006, 6: 13F-25F.
[10] Mallilankaraman K, Cardenas C, Doonan PJ, et al. MCUR1 is an essential component of mitochondrial Ca2+uptake that regulates cellular metabolism. Nat Cell Biol, 2012, 14: 1336-1343.
[11] Dong Z, Saikumar P, Weinberg JM, et al. Calcium in cell injury and death. Annu Rev Pathol, 2006, 1: 405-434.
[12] Foskett JK, Philipson B. The mitochondrial Ca(2+) uniporter complex. J Mol Cell Cardiol, 2015, 78: 3-8.
[13] 荆哲, 刘峰舟, 王炜中, 等. EMRE对高糖高脂诱导心肌细胞凋亡的影响. 心脏杂志, 2016, 3: 285-288.
[14] Liu T, Takimoto E, Dimaano VL, et al. Inhibiting mitochondrial Na+/ Ca2+exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res, 2014. 115: 44-54.
Preliminary Research for the Effect of EMRE on Myocardial Ischemia Injury in Experimental Mice
LIU Feng-zhou, JING Zhe, LIU Ming-li, ZHANG En-wei, NING Jiang-hua, MOU Jiao.
Department of Cardiology, Xijing Hospital of Forth Military Medical University, Xi’an (710032), Shaanxi, China
MOU Jiao, Email: fzzxmj@126.com
Objective: To study the effect of essential MCU regulator (EMRE) on myocardial ischemia injury in experimental mice with underlining mechanism.
Methods: Myocardial EMRE expression was up-regulated by EMRE adenovirus (Ad-EMRE) injection in mice myocardium tissue. Our research included in 3 groups: Sham operation group, sham mice received myocardium injection of eGFP adenovirus (Ad-eGFP); Myocardial infarction (MI) control group, the mice received Ad-eGFP injection and 48 hours later had coronary LAD ligation to establish MI model; MI treatment group, MI mice received Ad-EMRE injection. All animals were treated in 3 weeks. Mice cardiac function was examined by ultrasound; cardiomyocyte hypertrophy was evaluated by wheat germ agglutinin (WGA) staining, collagen fibrosis was measured by Masson staining, cell apoptosis was determined by TUNEL assay, protein expressions of EMRE, caspase-3 and caspase-9 were detected by Western blot analysis and mitochondrial reactive oxygen species (ROS) was assayed by MitoSOX fluorescence probe.
Results: Compared with MI control group, MI treatment group showed the worse cardiac function, aggravated cardiac hypertrophy and elevated collagen fibrosis; in addition, MI treatment group had obviously increased cardiomyocyte apoptosis and increased protein expressions of Caspase-3, Caspase-9 and more mitochondrial ROS production.
Conclusion: Over expressed EMRE can increase mitochondrial ROS production, induce cardiomyocyte apoptosis and therefore, aggravate myocardial ischemia injury in experimental mice.
Mitochondrial transporter proteins; Myocardial ischemia
2016-11-29)
(编辑:许菁)
陕西省自然科学基础研究计划资助项目(2015JM8479)
710032 陕西省西安市,中国人民解放军第四军医大学第一附属医院 心血管内科(刘峰舟、荆哲);中国人民解放军第四军医大学唐都医院 骨科(张恩尉);西安市中心医院(牟佼);空军94195部队卫生队(刘峰舟、宁江华);解放军兰州总医院 心内科(荆哲);宝鸡市第二中医医院 呼吸内分泌科 (刘明莉)
刘峰舟 硕士研究生 主要研究方向:糖尿病心肌病及心肌缺血损伤防治 Email: liufengzhou1986@163.com 通讯作者:牟佼
Email: fzzxmj@126.com
R54
A
1000-3614(2017)07-0701-06
10.3969/j.issn.1000-3614.2017.07.019
