赖氨酸尿性蛋白耐受不良1家系(1例合并系统性红斑狼疮)报告并文献复习
2017-07-31李国民刘海梅周利军吴冰冰冯佳燕
李国民 刘海梅 张 涛 史 雨 姚 文 周利军 徐 虹 吴冰冰冯佳燕 陆 炜 孙 利
·论著·
赖氨酸尿性蛋白耐受不良1家系(1例合并系统性红斑狼疮)报告并文献复习
李国民1)5 )刘海梅1)5)张 涛1)史 雨1)姚 文1)周利军1)徐 虹1)吴冰冰2)冯佳燕3)陆 炜4)孙 利1)
目的 总结赖氨酸尿性蛋白耐受不良(LPI)1家系2例患者临床特征及基因突变的特点,提高对该病的认识。方法 收集PLI 1家系2例患儿(同胞姐弟)的临床资料,包括病史、肾脏病理、相关实验室检查和家族史等。采用外显子捕获法对患儿及其父母行全外显子测序(WES)并行生物信息学分析,行Sanger验证并在家系其他成员中进行突变分析。结果 先证者,女,10岁,断乳后反复呕吐和腹泻,抵触富含蛋白的食物,生长落后,身高低于同年龄、同性别儿童;5岁后鼻自发性出血,外周血“三系”(RBC、WBC、PLT)低于正常值,9.7岁后出现轻度蛋白尿和持续性镜下血尿;免疫系统受累,检出ANA和ds-DNA等多种自身抗体。血清铁蛋白、乳酸脱氢酶和血氨增高,尿乳清酸增高,血清和尿液赖氨酸、精氨酸和瓜氨酸改变不明显。肾活检病理提示狼疮性肾炎。家系调查发现,先证者之弟,男,6.5岁,断乳后反复呕吐,抵触富含蛋白食物,生长落后,身高低于同年龄、同性别儿童,血清铁蛋白、乳酸脱氢酶增高,尿乳清酸增高,血清、尿液赖氨酸、精氨酸和瓜氨酸改变不明显。先证者及其父母WES测序显示SLC7A7基因IVS4+1G>A纯合突变,突变来源其父母,先证者之弟也存在该突变。例1诊断为LPI合并SLE,例2诊断为LPI。结论SLC7A7基因测序是确诊LPI的依据;LPI并发系统性红斑狼疮(SLE)非常罕见,SLC7A7是否为单基因型SLE致病基因需进一步研究。
赖氨酸尿性蛋白耐受不良; 赖氨酸; 系统性红斑狼疮;SLC7A7基因
1 病例资料
例1(先证者)女,2006年9月11日出生,2016年10月15日因“反复鼻出血、PLT低于正常值5年、反复发热3月”就诊复旦大学附属儿科医院(我院),拟以系统性红斑狼疮(SLE)收入我院。
患儿5年前(2011年10月)无明显诱因鼻出血,就诊当地医院,血常规提示PLT低于正常值(具体数值不详),予“地榆生白片、利可君片”等口服治疗2周。随后4年半内,每年3~4次鼻出血,未经特殊处理自行缓解。半年前鼻出血加重,出血时间延长,就诊当地医院,血常规提示WBC、RBC和PLT(简称“三系”)均低于正常值,骨髓穿刺提示增生性贫血、吞噬网状细胞1%,骨髓活检提示骨髓增生活跃,粒红比例大致正常,巨核细胞可见,分化欠成熟。未予特殊治疗。3个月前不规则发热,最高体温40℃(肛温),院外抗生素治疗5 d后体温恢复正常,5~6 d后再次发热,经抗生素治疗体温仍可恢复正常,反复发热3次,期间多次血常规提示“三系”低于正常值,尿常规蛋白定性-至++,持续镜下血尿(60.8~122.7个/HP)。1周前再次发热,热出疹出,热退疹退,伴有左膝关节疼痛。3 d前就诊我院门诊,抗核抗体(ANA)、抗双链DNA(ds-DNA)抗体1∶101阳性、抗SSA抗体阳性。
患儿系G1P1,足月顺产,出生体重3 300 g,出生时无产伤和窒息。母乳喂养4个月,断乳后反复呕吐和腹泻,每次持续3~5 d,间隔时间无规律性,抵触富含蛋白食物。父母体健,非近亲结婚。母亲孕产史为2-0-0-2,弟弟5岁(例2)。
查体:血压110/85 mmHg,身高126 cm(-2.6 SD),体重22 kg,神志清楚,精神一般,轻度贫血貌,无皮疹,营养状况尚可,未触及浅表淋巴结肿大,心脏和肺听诊未发现异常,腹软、平坦、无压痛和反跳痛,肝、脾肋下触未及,神经系统检查未见异常。
入院后多次血常规示“三系”低于正常值(表1)、尿沉渣检查蛋白质微量至+,镜下血尿,RBC信息非均一性、RBC畸形率70%~80%,24 h尿蛋白定量0.37 g,α1微球蛋白(A1MU)/肌酐(Cr)17.7 mg·g-1、白蛋白/Cr 300.7 mg·g-1、球蛋白(IGGU)/Cr 40.0 mg·g-1、N-乙酰-β-氨基葡萄糖苷酶(NAG) /Cr 2.83 U·mol-1、尿转铁蛋白3.4 mg·L-1,提示肾小球性蛋白异常。肝、肾功能指标和补体均在正常范围;免疫球蛋白IgA和IgG增高,余在正常范围;铁蛋白和乳酸脱氢酶增高(表1)。自身抗体ANA、抗ds-DNA抗体、抗SSA抗体、抗核小体抗体和抗核糖核蛋白抗体均阳性。血、尿串联质谱显示赖氨酸、精氨酸和瓜氨酸均在正常值范围(未年龄标化),尿串联质谱显示乳清酸浓度增高。肾组织取活检,光镜下可见15个肾小球,半数肾小球有轻度系膜细胞增生伴间质增多,其中2个肾小球内纤维新月体形成,个别肾小球毛细血管襻与球囊粘连;近端小管空泡样变性,个别小管灶性坏死伴淋巴细胞浸润,管腔内见少量红细胞管型、蛋白管型和细胞管型;肾间质见广泛分布的淋巴细胞、浆细胞浸润伴轻度纤维化,多个血管内膜轻度纤维性增厚。免疫荧光(图1A)显示IgG(-)、IgA(++)、IgM(±)、C3(+)、C1q(+)、Fb(+)。系膜细胞和间质轻度(图1B)至中度增生伴纤维新月体形成(图1C)。电镜(图1D)显示系膜区中等致密物沉积、内皮下和上皮下少量致密物沉积,系膜细胞和间质轻度增生,足细胞足突部分融合。肾脏病理表现符合狼疮性肾炎Ⅲ。腹部B超提示肾、肝和脾无明显异常。骨密度Z值-2.4(正常值-1~1)。
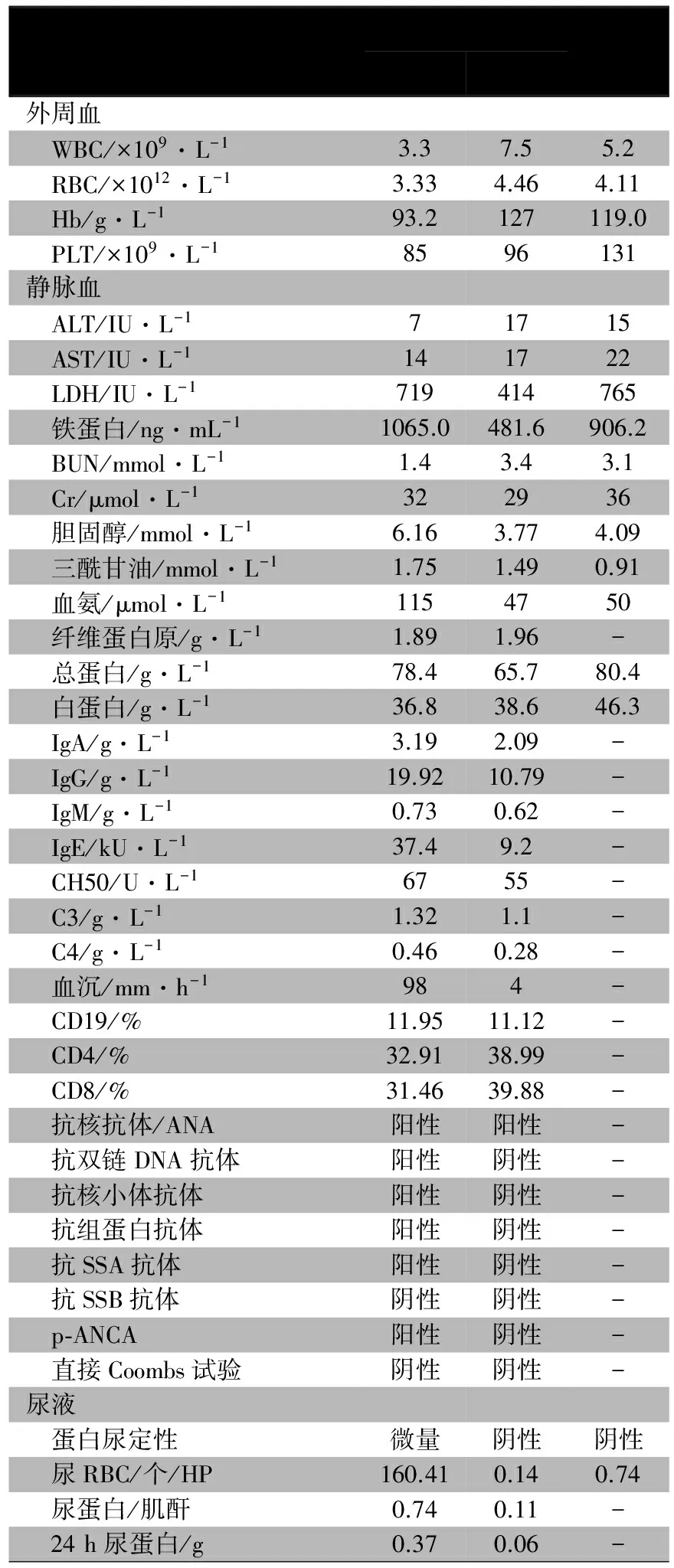
表1 先证者及其弟弟的实验室检查结果
注 -:未检测
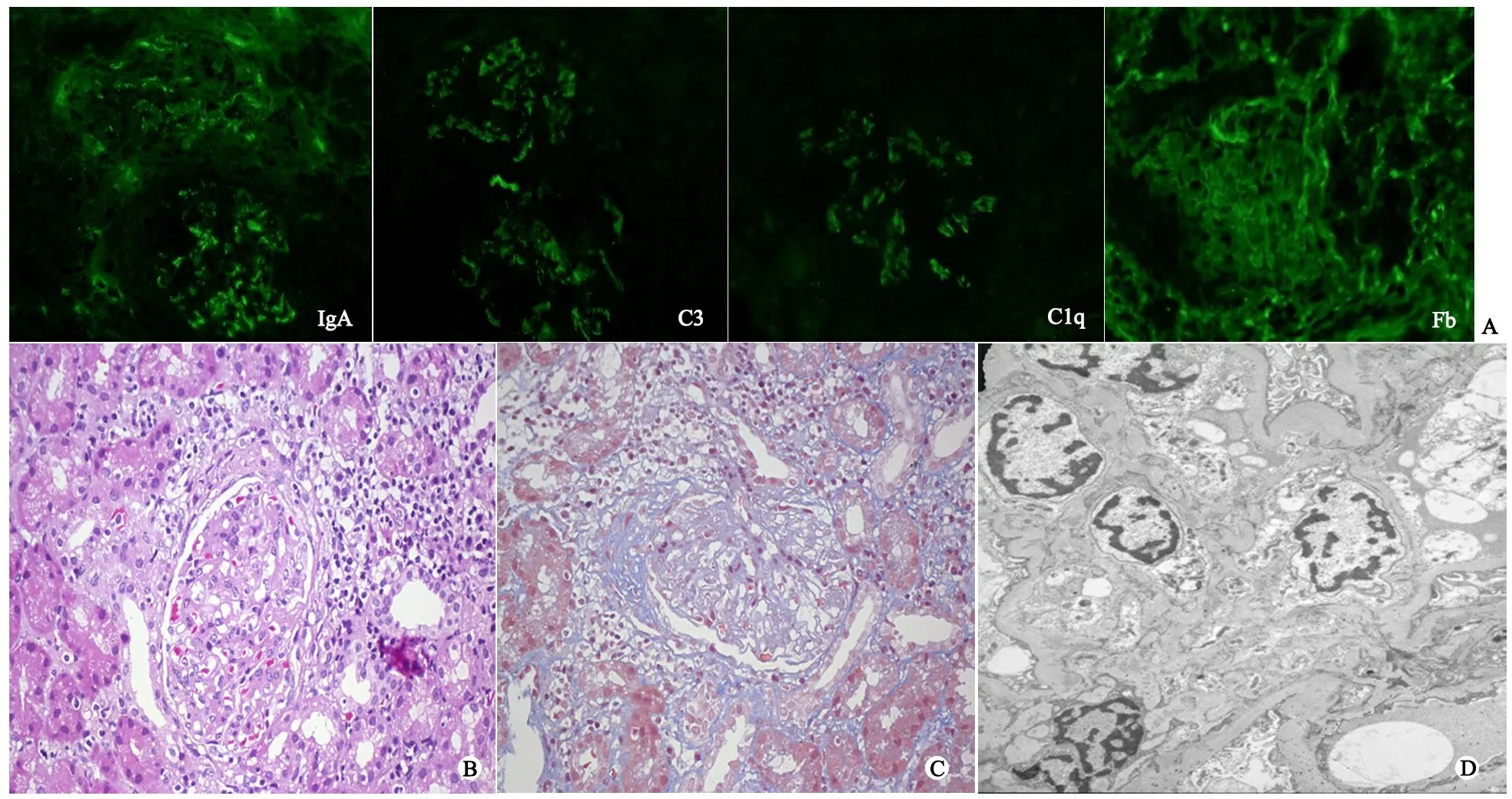
图1 肾组织镜下观察和免疫荧光
注 A:免疫荧光,IgA(++)、C3(+)、C1q(+)、Fb(+);B:HE染色(×200),系膜细胞和间质轻度增生;C:MAS染色(×200),系膜细胞和间质中度增生伴纤维新月体形成;D:电镜 (×11 500)系膜区中等致密物沉积、内皮下和上皮下少量致密物沉积,系膜细胞和间质轻度增生
例2(先证者之弟)男,2011年1月11日出生,2017年2月23日曾因“身材矮小”就诊于我院内分泌科,入院时身高106.5 cm(-2.8 SD),肝脏右肋下3.0 cm,剑突下4.7 cm,未发现其他阳性体征。生长激素激发试验无异常,血、尿串联质谱结果同先证者,其他检查结果见表1。全身骨密度Z值-4.1,腹部B超提示肝肿大。经入院评估未能找到身材矮小原因。出生至今有反复呕吐史,多于进食后,无发作性腹泻,不喜富含蛋白食物,曾于4和8月龄诊断“肺炎”,2岁时诊断“心肌炎”。
依据2例患儿的临床表现和实验室指标,临床诊断氨酸尿性蛋白耐受不良(LPI),予低蛋白饮食(1 g·kg-1·d-1)、瓜氨酸(100 mg·kg-1·d-1)分2次口服,随访血氨浓度均在正常范围。因全身骨密度均较低,给予钙剂和活性VD3口服。先证者还予泼尼松龙和霉酚酸酯治疗SLE,随访尿常规、补体正常,抗ds-DNA阴性,血常规除PLT计数偏低,WBC和RBC计数均在正常范围。
2 测序分析
2.1 全外显子测序(WES) 因先证者既往反复呕吐和腹泻、身材矮小、抵触富含蛋白食物等均无法用SLE来解释,取得患儿父母知情同意后对患后及其父母进行WES,方法如下。取静脉血2 mL,置EDTA抗凝管中混匀,用QIAamp DNA Mini试剂盒(美国Qiagen公司)提取基因组DNA,Nanodrop分光光度仪(美国Thermo公司)测定DNA浓度,TruSeqTM外显子富集试剂盒(美国Illumina公司)捕获外显子,按照Hiseq2500标准流程进行PE100测序,依照Q20标准进行测序结果筛选。
测序原始数据使用Burrows-Wheeler Aligner(BWA)、Picard、GATK在线软件进行初步数据分析,得到变异位点共63 219个。采用我院已建立的WES数据分析流程,逐步筛选,得到92个变异位点,根据先证者表型进一步人工筛选。
检测到符合先证者主要临床表型及遗传模式的可能致病基因1个,即SLC7A7基因经典剪切区IVS4+1G>A纯合变异,来源于先证者父母。该变异为HGMD数据库已报道的致病性突变。
2.2 Sanger 测序 采用Bigdye3.0(美国ABI公司)、3500xl
DNA Analyzer(美国 Thermo Fisher)对高通量测序结果进行Sanger验证,测序结果采用Mutation Surveyor(V4.0.9,美国SoftGenetics公司)分析,证实WES的SLC7A7基因在该家系的测序结果,先证者弟弟与先证者有相同基因型(图2)。
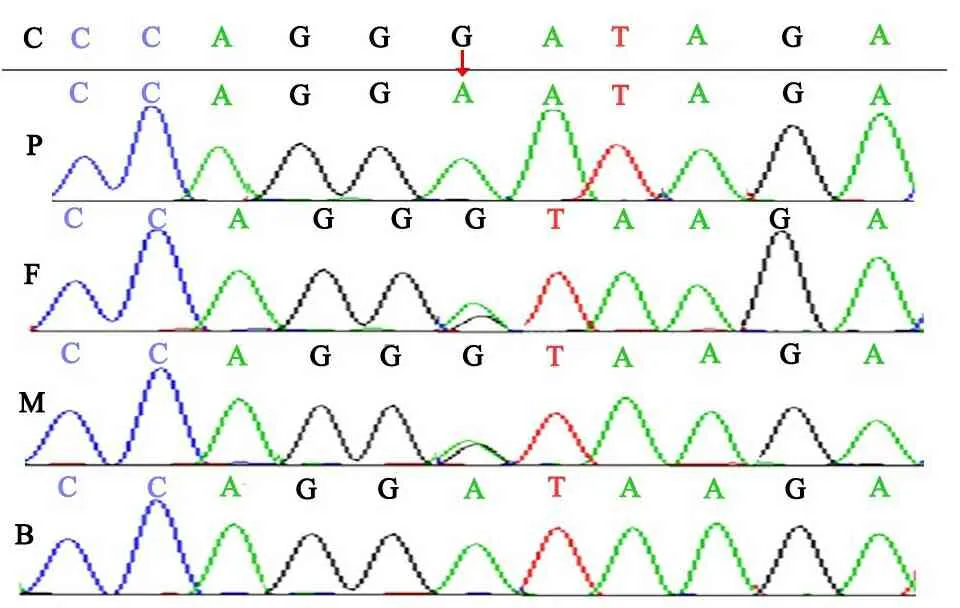
图2 高通量测序结果图
注SLC7A7基因 IVS4+1G>A突变家系验证,C:对照,P:先证者,F:先证者之父,M:先证者之母,B先证者之弟,红色箭头表示突变位置
3 文献检索和复习


表2 LPI临床主要并发症和异常实验室指标
注 括号中斜线后为报告例数,斜线前为阳性例数

表3 8 例LPI患者并发SLE的临床特征
注 N:资料不详
4 讨论
本文中先证者的临床表现及相关实验室检查结果符合SLE诊断标准,包括①血液系统受累,表现为Hb、WBC和PLT均低于正常值,且符合SLE血液系统受累的标准[19];②肾脏受累,表现为持续性镜下血尿,伴蛋白尿,24 h尿蛋白定量0.37 g(>0.150 g),符合SLE肾脏受累的儿童标准[20],肾活检提示狼疮性肾炎Ⅲ;③免疫学指标ANA和抗ds-DNA抗体阳性。患儿4月龄断母乳后至今反复呕吐、腹泻,每次3~5 d,间隔时间无规律性;入院时身高126 cm(-2.6SD);不喜富含蛋白食物,均无法用SLE来解释。此外,经泼尼松龙和霉酚酸酯口服治疗SLE,尿蛋白转阴,镜下血尿消失,外周血“三系”恢复正常,但LDH和铁蛋白等生化指标仍然异常,也无法用SLE来解释。血、尿串联质谱显示氨基酸均在正常值范围,而尿乳清酸增高明显,不排除遗传代谢性疾病可能。予先证者及其父母WES,结果显示,患儿存在SLC7A7基因IVS4+1G>A纯合突变,该突变为经典的剪切位点突变,为已报道致病性突变。SLC7A7基因突变可引起LPI。
LPI(OMIM #222700)是一种非常罕见的先天性代谢性疾病,为常染色体隐性遗传[1, 21]。1965年,该病首先由芬兰学者报告,世界各地散发,法国发病率为1.7/10万活产新生儿,芬兰为1/6万活产新生儿,日本为1/5.7万活产新生儿[9, 22~24]。LPI由SLC7A7基因突变引起,该基因编码y+LAT1蛋白,为双碱基氨基酸转运体的一个轻链亚基,主要在小肠黏膜、肾小管和肝脏等表达[25]。缺陷的双碱基氨基酸转运体不能将肠道的赖氨酸、精氨酸和鸟氨酸转运至体内,造成血清上述3种阳离子氨基酸浓度低于正常,肾小管不能重吸收这些氨基酸,引起这些氨基酸随尿液排出增多,从而使肝细胞缺乏足够的底物参与尿素循环,继发尿素循环障碍,触发高氨血症[26,27]。LPI特征性表现为:母乳喂养或混合喂养后发作性呕吐和腹泻、高蛋白饮食后的精神错乱和昏迷、喂养困难、抵触富含蛋白的食物、生长迟缓、肝脾肿大和肌力减退[28,29]。随着时间推移,除了以上非特异性临床表现外,还会累及其他系统,出现多个系统并发症,但并非每个患者均有上述所有并发症。LPI患者可出现多项实验室指标异常,概括为两类,一类为双碱基氨基酸转运障碍和继发尿素循环障碍所引起,如血清赖氨酸、精氨酸和鸟氨酸浓度下降,尿中上述氨基酸排泄增多,血氨浓度上升;另一类为亚临床巨噬细胞活化指标,如外周血血细胞计数下降,转氨酶、乳酸脱氢酶、铁蛋白和三酰甘油增高以及凝血功能异常[3,27]。
LPI诊断主要依据临床表现和实验室指标,早期临床表现出现在断乳后,为非特异性,容易漏诊或误诊,往往在出现其他系统并发症后才引起患儿家长和医生的重视,行血、尿氨基酸测定和血氨等生化检测。尽管LPI出生后就起病,由于早期症状不典型,部分病例到成人期才被诊断。本文先证者,出现血液系统“三系”低于正常值,蛋白尿、血尿等肾脏受累表现,自身抗体ANA、抗ds-DNA抗体阳性,诊断SLE后才引起临床的重视。例2主要表现为身材矮小、肝脏肿大,反复呕吐(可自然缓解),不喜富含蛋白的食物,由于对LPI认识不足,以“矮小症”收住入院。2例患儿血、尿串联质谱结果提示赖氨酸、精氨酸和鸟氨酸浓度均在“正常值范围”,这给临床诊断造成困难。虽然实验室检查是LPI的重要诊断依据,由于两类实验室指标无直接相关性,特别是亚临床巨噬细胞活化指标很难用原发疾病解释,该类指标临床中容易被忽视,而双碱基氨基酸转运障碍指标受人为因素的影响,也易被误判为“正常”。造成这一现象可能与该“正常值范围”没有用年龄进行标化有关。
本文先证者及其父母行WES,发现存在SLC7A7基因IVS4+1G>A纯合突变,该突变来源于其父母,Sanger法验证了该突变,为已报道的致病性突变,且例2与先证者具有相同基因型。测序结果进一步证实患儿LPI诊断。SLC7A7基因IVS4+1G>A突变多见亚洲人[4]。本文2例基因型相同但临床表型不尽相同,进一步提示LPI的临床异质性。目前关于PLI病例报告主要见于国外,国内仅见1例成人报告[2]。
LPI并发SLE十分罕见,截至目前全球仅有8例报告(含本例)[12~18],除本研究先证者外,其他7例均未行SLC7A7基因检测。多数学者认为,GC治疗是必要的,并且时间≥6个月[15~18]。8例患儿中4例死亡,其中例2~5均接受GC治疗,但均死于肺部并发症,肺部受累可能是造成PLI并发SLE患儿最主要死亡原因。LPI并发SLE机制尚不清楚,巨噬细胞和淋巴细胞均有y+LAT1蛋白表达,SLC7A7基因突变引起以上细胞中氨基酸代谢异常,导致固有免疫和适应性免疫细胞能量代谢异常,从而导致免疫耐受破坏,出现自身抗体[11,30,31]。
SLE由环境因素、免疫功能异常和遗传因素共同引起。然而,少部分SLE病例是由C1q、C1r、C1s、C2、C3、C4A、Dnase1、DNase1L3、ACP5、PRKCD、IFIH1、TREX-1和SAMHD1等单基因突变引起[32~34],尚未有报道将SLC7A7基因作为单基因型SLE的致病基因。由于SLC7A7基因突变不仅仅引起LPI,同时可以伴发SLE,可能为单基因型SLE的致病基因。
LPI治疗一是减少高氨血症所致风险,提供相对足够蛋白和必须氨基酸维持自然生长;二是相关并发症的治疗[ 35,36]。减少高氨血症所致风险主要措施有低蛋白饮食和瓜氨酸替代治疗,血氨浓度控制在正常范围内后可适当增加蛋白摄入,促进生长;出现并发症应给予适当治疗,如PLI并发间质性肺炎、噬血细胞综合征和SLE等,需GC或GC联合免疫抑制剂治疗。LPI患者一般都有骨密度减低,进一步会发展为骨质疏松症,甚至导致多发骨折,早期予活性维生素D和钙剂,必要时予阿仑磷酸盐口服,可以预防骨质疏松和骨折发生[37]。早期诊断、合理干预可有效减少并发症特别是严重并发症的出现,改善预后。
[1] Torrents D, Mykkänen J, Pineda M, et al. Identification of SLC7A7, encoding y+LAT-1, as the lysinuric protein intolerance gene. Nat Genet,1999, 21(3):293-296
[2] 彭方,陆珺,张祥,等. 赖氨酸尿性蛋白耐受不良一例. 中华神经科杂志. 2016,49(11):874-876
[3] Valimahamed-Mitha S, Berteloot L, Ducoin H, et al. Lung involvement in children with lysinuric protein intolerance. J Inherit Metab Dis, 2015, 38(2): 257-263
[4] Tanner LM, Näntö-Salonen K, Niinikoski H, et al. Nephropathy advancing to end-stage renal disease: a novel complication of lysinuric protein intolerance. J Pediatr, 2007, 150(6):631-634
[5] Parenti G, Sebastio G, Strisciuglio P, et al. Lysinuric protein intolerance characterized by bone marrow abnormalities and severe clinical course. J Pediatr. 1995, 126(2):246-251
[6] Svedström E, Parto K, Marttinen M, et al. Skeletal manifestations of lysinuric protein intolerance. A follow-up study of 29 patients. Skeletal Radiol, 1993, 22(1): 11-16
[7] Güzel-Ozantürk A, Ozgül RK, Unal O, et al. Molecular and clinical evaluation of Turkish patients with lysinuric protein intolerance. Gene, 2013, 521(2):293-295
[8] Parto K, Penttinen R, Paronen I, et al. Osteoporosis in lysinuric protein intolerance. J Inherit Metab Dis, 1993, 16(2): 441-450
[9] Noguchi A, Nakamura K, Murayama K, et al. Clinical and genetic features of lysinuric protein intolerance in Japan. Pediatr Int, 2016, 58(10): 979-983
[10] Reinoso MA, Whitley C, Jessurun J, et al. Lysinuric protein intolerance masquerading as celiac disease: a case report. J Pediatr, 1998, 132(1): 153-155
[11] Lukkarinen M, Parto K, Ruuskanen O, et al. B and T cell immunity in patients with lysinuric protein intolerance. Clin Exp Immunol, 1999, 116(3):430-434
[12] Nagata M, Suzuki M, Kawamura G, et al. Immunological abnormalities in a patient with lysinuric protein intolerance. Eur J Pediatr, 1987, 146(4):427-428
[13] Parto K, Kallajoki M, Aho H, et al. Pulmonary alveolar proteinosis and glomerulonephritis in lysinuric protein intolerance: case reports and autopsy findings of four pediatric patients. Hum Pathol, 1994, 25(4): 400-407
[14] Di Rocco M, Buoncompagni A, Gattorno M, et al. Complications of lysinuric protein intolerance must be treated with immunosuppressive drugs. J Inherit Metab Dis, 1998, 21(6):675-676
[15] Parsons H, Snyder F, Bowen T, et al. Immune complex disease consistent with systemic lupus erythematosus in a patient with lysinuric protein intolerance. J Inherit Metab Dis, 1996, 19(5):627-634
[16] Kamoda T, Nagai Y, Shigeta M, et al. Lysinuric protein intolerance and systemic lupus erythematosus. Eur J Pediatr, 1998, 157(2):130-131
[17] Aoki M, Fukao T, Fujita Y, et al. Lysinuric protein intolerance in siblings: complication of systemic lupus erythematosus in the elder sister. Eur J Pediatr. 2001, 160(8):522-523
[18] Nicolas C, Bednarek N, Vuiblet V, et al. Renal involvement in a French paediatric cohort of patients with lysinuric protein intolerance. JIMD Rep, 2016, 29(1):11-17
[19]Petri M, Orbai AM, Alarcón GS,et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum, 2012, 64(8):2677-2686
[20]中华医学会儿科分会肾脏病学组.狼疮性肾炎诊疗指南.中华儿科杂志, 2010, 48(9):687-689
[21] Mauhin W, Habarou F, Gobin S, et al. Update on lysinuric potein intolerance, a multi-faceted disease retrospective cohort analysis from birth to adulthood. Orphanet J Rare Dis, 2017, 12(1):3
[22] Estève E, Krug P, Hummel A, et al. Renal involvement in lysinuric protein intolerance: contribution of pathology to assessment of heterogeneity of renal lesions. Hum Pathol, 2017,62(1):160-169
[23] Noguchi A, Nakamura K, Murayama K, et al. Clinical and genetic features of lysinuric protein intolerance in Japan. Pediatr Int, 2016, 58(10):979-983
[24]Koizumi A, Shoji Y, Nozaki J, et al. A cluster of lysinuric protein intolerance (LPI) patients in a northern part of Iwate, Japan due to a founder effect. The Mass Screening Group. Hum Mutat, 2000, 16(3):270-271
[25] Borsani G, Bassi MT, Sperandeo MP, et al. SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat Genet, 1999,21(3):297-301
[26] Mykkänen J, Torrents D, Pineda M, et al. Functional analysis of novel mutations in y(+)LAT-1 amino acid transporter gene causing lysinuric protein intolerance (LPI). Hum Mol Genet, 2000, 9(3): 431-438
[27]Boenzi S, Pastore A, Martinelli D, et al. Creatine metabolism in urea cycle defects. J Inherit Metab Dis, 2012, 35(4): 647-653
[28] Sebastio G, Sperandeo MP, Andria G. Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am J Med Genet C Semin Med Genet, 2011, 157C:54-62
[29] Bröer S. Lysinuric protein intolerance: one gene, many problems. Am J Physiol Cell Physiol, 2007, 293(2):C540-541
[30] Kurko J, Vähä-MäkiläM, Tringham M, et al. Dysfunction in macrophage toll-like receptor signaling caused by an inborn error of cationic amino acid transport. Mol Immunol, 2015, 67(2 Pt B):416-425
[31] Barilli A, Rotoli BM, Visigalli R, et al. Impaired phagocytosis in macrophages from patients affected by lysinuric protein intolerance. Mol Genet Metab, 2012, 105(4): 585-589
[32] Volpi S, Picco P, Caorsi R, et al. Type I interferonopathies in pediatric rheumatology. Pediatr Rheumatol Online J, 2016,14(1): 35
[33] Chen L, Morris DL, Vyse TJ. Genetic advances in systemic lupus erythematosus: an update.Curr Opin Rheumatol, 2017 [Epub ahead of print][34]Teruel M, Alarcón-Riquelme ME. The genetic basis of systemic lupus erythematosus: What are the risk factors and what have we learned. J Autoimmun, 2016, 74:161-175
[35] Lukkarinen M, Nanto-Salonen K, Pulkki K, et al. Oral supplementation corrects plasma lysine concentrations in lysinuric protein intolerance. Metabolism, 2003, 52: 935-938
[36] Dionisi-Vici C, De Felice L, el Hachem M, et al. Intravenous immune globulin in lysinuric protein intolerance. J Inherit Metab Dis, 1998, 21(2): 95-102
[37] Posey JE, Burrage LC, Miller MJ, et al. Lysinuric Protein Intolerance Presenting with Multiple Fractures. Mol Genet Metab Rep, 2014, 1: 176-183
(本文编辑:张崇凡,孙晋枫)
Lysinuricproteinintoleranceinsiblings:complicatedwithsystemiclupuserythematosusintheeldersisterandliteraturereview
LIGuo-min1)5),LIUHai-mei1)5),ZHANGTao1),SHIYu1),YAOWen1),ZHOULi-jun1),XUHong1),WUBing-bing2),FENGJia-yan3),LUWei4),SUNLi1)
(Children'sHospitalofFudanUniversity,Shanghai201102,China; 1)DepartmentofRheumatology, 2)MedicalTranslationalCenter, 3)DepartmentofPathology, 4)DepartmentofEndocrinology, 5)Co-firstauthor)
Corresponding Author:SUN Li, E-mail: lillysun@263.net
ObjectiveTo summarize and review the clinical data of two children with lysinuric protein intolerance so as to improve its knowledge. MethodsClinical data of two cases with lysinuric protein intolerance were summarized, including clinical manifestations, laboratory findings, renal pathological changes and family investigation. This study used next generation sequencing to screen all exons of genome in proband and her parents. Significant variants detected by next generation sequencing were confirmed by conventional Sanger sequencing and segregation analysis was performed using parental DNA and her brother samples.ResultsThe proband, a 10-year-old girl, presented with recurrent vomiting and episodes of diarrhea, aversion to protein-rich food and failure to thrive after weaning. She often had nasal hemorrhage since the age of 5 years. Peripheral blood cell count suggested white blood cell, red blood cell and platelet count were all under normal value. She had mild proteinuria and persistent microscopic hematuria at the age of 9.7 years. At the same time, laboratory tests showed that serum ferritin, lactate dehydrogenase and ammonia increased, and orotic acid increased in urine, but lysine, arginine and citrulline were not changed significantly in serum and urine. The pathology of renal biopsy suggested lupus nephritis. The proband's younger brother, 6.5-year-old, presented with recurrent vomiting, aversion to protein-rich food and failure to thrive after weaning. He had no episodes of diarrhea. laboratory tests also showed that serum ferritin, lactate dehydrogenase and ammonia increased, and orotic acid increased in urine, but lysine, arginine and citrulline were not changed significantly in serum and urine. Whole exon sequencing was performed in core family, including proband and her parents. Homozygous c.625+1G>A mutation inSLC7A7 gene was detected in proband, which was from her parents. The mutation was confirmed by Sanger sequencing in core family. The same mutation was found in proband's younger brother by Sanger sequencing. The proband was diagnosed as LPI complicted with SLE. The proband's younger brother was diagnosed as LPI.ConclusionDue to the heterogeneity of LPI and lack of understanding of LPI for clinicians, it is easy to cause misdiagnosis or miss diagnosis. TheSLC7A7 gene sequencing is the basis for diagnosis. LPI patients with systemic lupus erythematosus (SLE) is very rare. LPI patients complicated with SLE need glucocorticoid or immunosuppressive therapy. Mutations inSLC7A7 gene can cause SLE. whetherSLC7A7 is one of the genes causing a single gene type SLE needs further study.
Lysinuric protein intolerance; Lysine; Systemic lupus erythematosus;SLC7A7 gene
1复旦大学附属儿科医院 上海,201102;1)风湿科,2)医学转化中心, 3)病理科,4)内分泌科,5)共同第一作者
孙利,E-mail:lillysun@263.net
10.3969/j.issn.1673-5501.2017.03.007
2017-06-12
2017-06-24)
