儿童慢性炎性脱髓鞘性多发性神经病15例报告并文献复习
2017-07-31柴毅明
邱 甜 柴毅明
·论著·
儿童慢性炎性脱髓鞘性多发性神经病15例报告并文献复习
邱 甜 柴毅明
目的 探讨儿童慢性炎性脱髓鞘性多发性神经病(CIDP)的临床特点和预后。方法 收集2008年1月至2017年1月复旦大学附属儿科医院收治的CIDP患儿的临床资料,检索PubMed数据库1991年1月至2017年2月的儿童CIDP病例,将文献检索和本文的病例汇总,根据到达疾病高峰期时间分组,4~8周为亚急性起病组,>8周为慢性起病组,比较两组的临床表现、实验室检查、治疗和预后。结果 ①符合本文纳入和排除标准的CIDP患儿15例,男6例,女9例;1~3岁4例,~7岁4例,~15岁7例;起病年龄1岁1月至11岁9月;7例有前驱感染史;到达疾病高峰期时间为4周至13月,平均(12.9±13.6)周。15例的首发症状均为运动障碍,4例有感觉障碍;疾病高峰期改良Rankin量表(MRS)评分:3分8例,4分6例,5分1例;病程呈单向型3例,进展型5例,复发型7例;15例神经传导检查均有神经源性损害,脑脊液检查均有细胞蛋白分离现象。14例予糖皮质激素治疗,1例予静脉丙种球蛋白治疗;1例家属放弃治疗出院,其余14例住院时间7~17 d,出院时临床症状均有不同程度好转。随访中7例患儿复发。②共有14篇文献报道87例CIDP患儿与本文15例合并行文献分析(n=102),亚急性起病组38例,慢性起病组64例。两组性别、起病年龄和运动障碍差异无统计学意义,感觉障碍(57.1%vs23.5%,P=0.002)和颅神经异常(25.7%vs7.8%,P=0.023)亚急性起病组高于慢性起病组,且更易反复发作(62.2%vs34.0%,P=0.010),慢性起病组单相发作形式多(50.8%vs27.8%,P=0.026)。结论 CIDP亚急性起病患儿感觉障碍和颅神经异常多于慢性起病患儿,并且疾病发展过程中容易复发。儿童CIDP运动障碍常见,较少出现颅神经损害和呼吸衰竭等,脑脊液细胞蛋白分离现象多见,电生理改变明显,早期治疗则预后较好。
慢性炎性脱髓鞘性多发性神经病; 临床分析; 儿童
慢性炎性脱髓鞘性多发性神经病(CIDP)是一种由免疫介导的慢性获得性运动感觉周围神经病,特点是慢性起病、四肢近端和远端对称性无力伴腱反射减弱和感觉异常[1]。儿童与成人CIDP相比发病率较低,但可遗留不同程度神经系统残疾,影响患儿的生活质量。对CIDP的早期诊断有助于预防神经系统后遗症,从而改善预后。本文回顾性收集复旦大学附属儿科医院(我院)近年来收治的CIDP患儿的临床资料并行文献复习,以提高对儿童CIDP临床特征的认识,促进早期诊断和治疗。
1 方法
1.1 CIDP诊断、治疗和随访方案 诊断参照第88届欧洲神经肌肉病中心(ENMC)会议修订的儿童CIDP诊断标准[2]和中国CIDP诊疗指南[3]。治疗:①糖皮质激素(首选),甲基泼尼松龙20 mg·kg-1·d-1,静脉滴注,3 d后改口服泼尼松2 mg·kg-1·d-1,维持4~6周后逐渐减量;或直接口服泼尼松2 mg·kg-1·d-1,维持4~6周后逐渐减量;口服泼尼松减量直至小剂量维持半年以上再酌情停药。②IVIG 400 mg·kg-1·d-1,静脉滴注,连续5 d为1个疗程;每月重复1次,连续3个月或以上。③血浆交换(有条件者),每个疗程3~5次,间隔2~3 d,每次交换量30 mL·kg-1,每月1个疗程。④上述治疗效果不理想或产生激素依赖或激素无法耐受,可选用或加用免疫抑制剂。要求出院后1个月内门诊随访1次,此后每3个月随访1次至停药,停药后每年电话随访1次。主要随访内容包括:颅神经、运动(肌力、肌张力、肌容积和腱反射)、感觉、日常生活能力、改良Rankin量表(MRS)[4]等。
1.2 相关分类和定义 ①CIDP患儿运动障碍和感觉异常的评价工具为MRS。0分:完全无症状;1分:尽管有症状,但无明显功能障碍,能完成所有日常活动;2分:轻度残疾,不能完成病前所有活动,但不需要帮助,能照顾自己的事务;3分:中度残疾,要求一些帮助,但行走不需要帮助;4分:重度残疾,不能独立行走,无他人帮助不能满足自身的需要;5分:严重残疾,卧床、失禁,要求继续护理。②4~8周到达疾病高峰期为亚急性起病,>8周为慢性起病[2];③疾病演变方式分单向型、进展型和反复型。单相型:病情单相进展后好转;进展型:病情逐渐加重,无明显好转期;反复型:在病程中有>2次不连贯的发病,中间有好转期。
1.3 病例纳入和排除标准 纳入2008年1月至2017年1月我院神经科收治的符合CIDP诊断标准的连续住院患儿,排除遗传性和药物或毒素导致的脱髓鞘性周围神经病。
1.4 截取资料 ①性别、发病年龄;②到达疾病高峰期的时间、前驱感染病史、急性期症状、疾病演变方式和高峰期MRS评分;③脑脊液生化和神经传导检查结果;④治疗情况;⑤随访和预后。
1.5 文献检索策略 以(CIDP OR "chronic inflammatory demyelinating polyneuropathy")AND(children)检索PubMed数据库,检索时间为1991年1月至2017年2月,纳入以第88届ENMC会议[2]、美国神经学会[5]或欧洲神经学会[6]CIDP诊断标准为诊断根据的文献,排除无详细临床资料、未描述到达疾病高峰期时间的文献。

2 结果
2.1 一般情况 符合本文纳入和排除标准的CIDP患儿15例,男6例,女9例;1~3岁4例,~7岁4例,~15岁7例;起病年龄1岁1月至11岁9月,平均5.8岁。15例患儿的临床资料见表1。
临床发现:在诊断CIDP前1月内7例有明确的感染病史,主要表现为上呼吸道感染症状(鼻塞、流涕、咳嗽和发热等)。15例到达疾病高峰期时间为4周至13月,平均(12.9±13.6)周。首发症状:15例均为运动障碍,其中13例为双下肢运动障碍,表现为独走容易摔跤,双下肢近端肌力Ⅴ-级、远端肌力Ⅳ级,肌张力无明显下降,腱反射减弱;1例表现为抬头无力、持物不稳和行走容易摔跤,颈部肌力Ⅳ级,四肢近端和远端肌力Ⅳ级,肌张力及腱反射无明显异常;1例为双上肢运动障碍,表现为持物不稳,双上肢近端肌力Ⅴ-级、远端肌力Ⅳ级,肌张力无明显下降,腱反射减弱。表1显示疾病高峰期临床进展情况:15例患儿的运动障碍均加重,MRS评分:3分8例,4分6例,5分1例。病程演变:单向型3例,进展型5例,复发型7例(复发2、3和4次者分别为4、2和1例)。
神经传导检查结果显示,15例均为神经源性损害,均在出现临床症状后4~12周内首次行脑脊液检查,脑脊液中细胞数、葡萄糖和氯化物均在正常范围,蛋白均增高[(470~3 402)mg·L-1],呈细胞-蛋白分离现象。
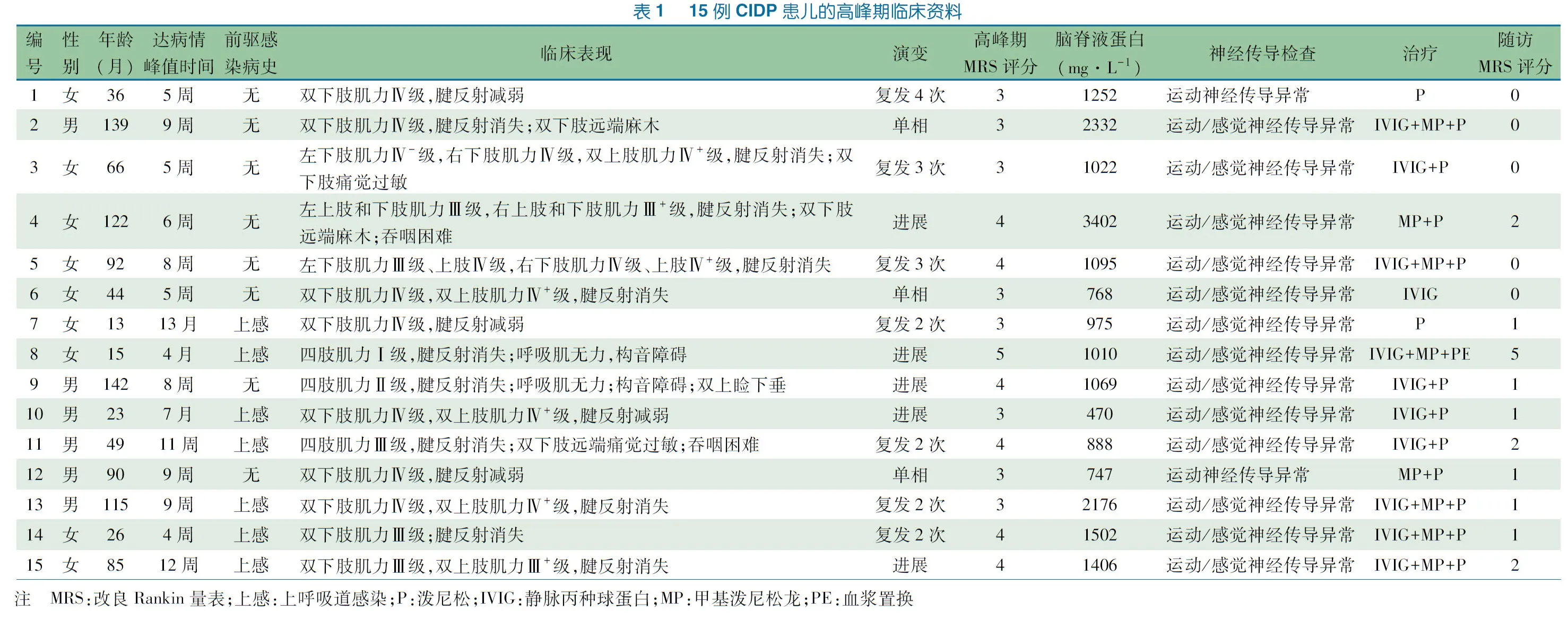
治疗及转归:14例患儿予糖皮质激素治疗,其中8例先予甲基泼尼松龙静脉滴注3 d,继以泼尼松口服治疗,6例予泼尼松口服治疗;8例同时予IVIG;泼尼松口服4~6周后,在6个月至1年内逐渐减量。1例患儿因家长拒绝使用糖皮质激素,予IVIG,持续5 d,每4周重复治疗1次,连续治疗4次。1例在病程中出现严重呼吸肌无力并发重症肺炎、呼吸衰竭,住院治疗9个月后,因呼吸机依赖患儿家属放弃治疗自动出院;其余14例(93%)住院时间7~17 d,出院时临床症状均有不同程度好转。
随访:7例复发。5例(33%)完全恢复;6例有下肢轻度疲劳,能独自完成所有日常活动,查体双侧下肢远端肌力Ⅴ-级;3例有下肢轻度远端无力,可以独走,上楼梯偶需辅助,查体双侧下肢远端肌力Ⅳ+级;1例放弃治疗、自动出院后严重残疾,长期卧床。
2.2 文献复习 14篇文献[7~20]符合本文设定的文献纳入和排除标准,共报道87例CIDP患儿,与本文报道的15例合并后共102例,亚急性起病组38例,慢性起病组64例。表2显示,两组性别、起病年龄和运动障碍方面差异无统计学意义,两组均无仅上肢受累的情况。亚急性起病组感觉异常(P=0.002)和颅神经异常(P=0.023)比例高于慢性起病组。与慢性起病组相比,亚急性起病组患儿的疾病演变过程更倾向于反复发作(P=0.010),慢性起病组单相型比例高于亚急性起病组(P=0.026)。两组中神经传导检查显示神经源性损害者和脑脊液>350 mg·L-1者均占90%以上,在神经传导检查的各项异常表现方面,两组差异均无统计学意义。两组的治疗方案、末次随访的MRS评分差异无统计学意义。
3 讨论
CIDP是一种获得性神经病变。目前关于CIDP的流行病学研究较少,有研究显示,成人CIDP患病率为(1~1.9)/100 000,儿童CIDP患病率0.48/100 000[21]。 由于CIDP临床症状隐匿且有异质性,非典型CIDP诊断困难,成人和儿童的CIDP患病率可能被低估。成人CIDP男性患病率较高,男女比例2.8∶1[22]。 本文病例男6例,女9例;文献复习结果中,男女比例差异无统计学意义。CIDP可在任何年龄发病,成人在40~60岁最常见[23]。本研究报道的15例儿童CIDP起病年龄1~11岁,平均5.8岁;文献复习结果中平均起病年龄7~8岁。
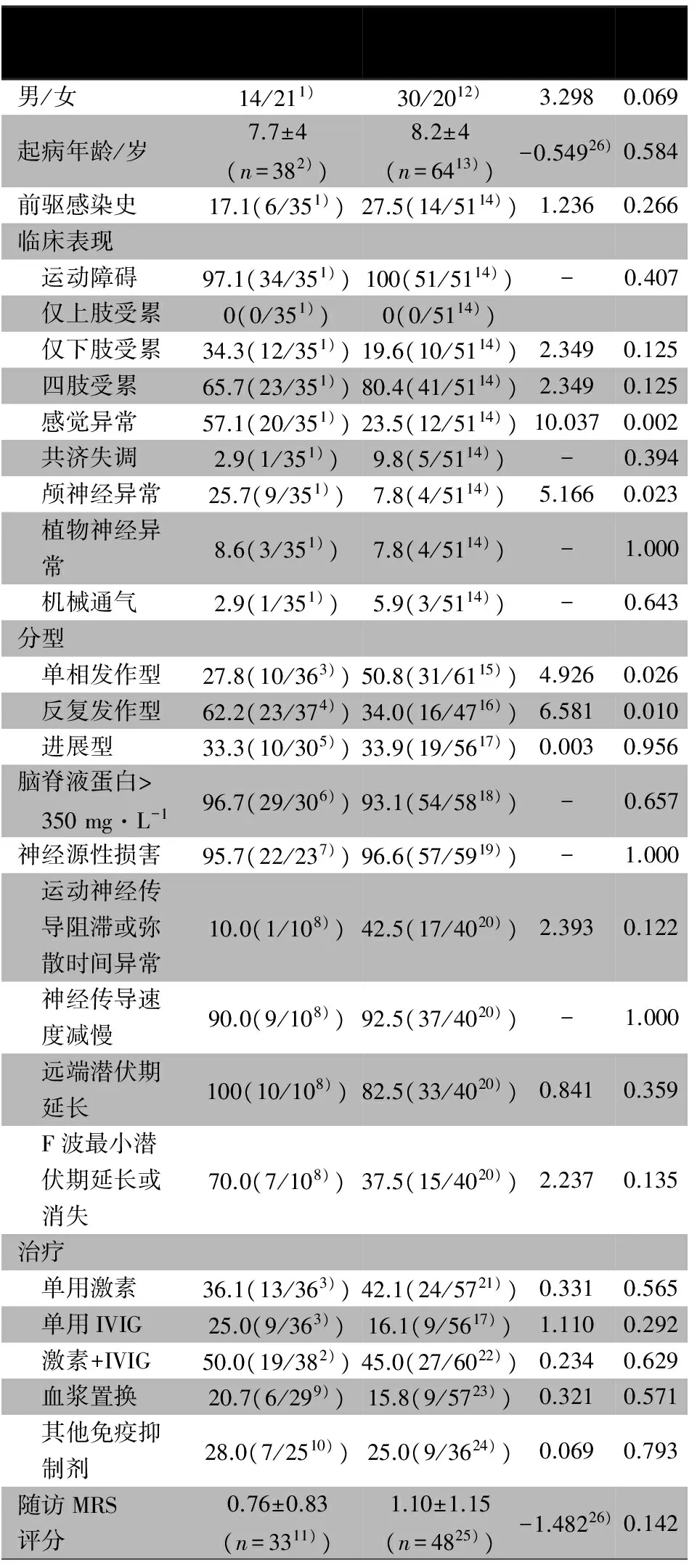
表2 亚急性起病组和慢性起病组对比分析[%(n/N)]
注 1)本文,文献7、9、13、14、16;2)本文,文献7~9、13、14、16;3)本文,文献7~9、16;4)本文,文献7~9、13、16;5)本文,文献7~9、14;6)本文,文献7、8、13、14、16;7)本文,文献7、8、13、14;8)本文,文献8、13、14;9)本文,文献7~9;10)7~9、14;11)本文,文献7、9、16;12)本文,文献7、9~11、13、15、17~20;13)本文,文献7~13、15、17~20;14)本文,文献7、9~13、15、17~20;15)本文,文献7~9、11、13、15、18~20;16)本文,文献7~10;17)本文,文献7~9、20;18)本文,文献7、8、10~13、15、17、19、20;19)本文,文献7、8、10~13、15、17~20;20)本文,文献8、10、11、13、15、17~20;21)本文,文献7~9、15、20;22)本文,文献7~9、11、13、18~20;23)本文,文献7~10、20;24)7~9;25)本文,文献7、9~11、15、18~20;26)t;余检验值为χ2
儿童CIDP最常见的主诉为行走困难和容易摔倒,甚至进展至失去行走的能力。除了双下肢运动功能障碍以外,还可以出现上肢无力、手震颤、腱反射减弱或者消失,或同时存在感觉障碍和感觉性共济失调[7]等。本文15例患儿均有运动障碍,受累部位以双下肢远端多见,其次为肢体远端和近端均等受累,绝大多数呈左右侧对称性受累,两侧肌力差距<1级;未见深感觉减退或感觉性共济失调,未发现明显自主神经功能障碍。
目前,CIDP缺乏特异性的生物学标志物,仍未统一临床诊断标准[24],儿童CIDP诊断主要依靠临床表现、神经电生理和脑脊液检查以及对治疗的反应性。第88届ENMC会议更新了儿童CIDP的诊断标准[2]。与儿不同,成人童CIDP诊断标准要求疾病症状进展>8周[3]。本文15例患儿均符合儿童CIDP诊断标准。文献复习中17例患儿符合美国神经学会[5]或欧洲神经学会[6]CIDP诊断标准;70例患儿符合第88届ENMC会议[2]诊断标准。 如患儿有CIDP的临床特征,即使不完全符合CIDP的诊断标准,也需要考虑CIDP诊断,早期给予干预,以免漏诊、误诊。
儿童CIDP治疗的主要目标是控制炎症以及防止发生轴索损害,主要治疗手段为免疫治疗,包括糖皮质激素、IVIG和血浆置换等。本文中通过糖皮质激素治疗患儿均在短期内有不同程度好转;例6仅使用IVIG治疗,完全缓解;例8行血浆置换治疗,但是由于患儿并发重症肺炎、呼吸衰竭,出现呼吸机依赖,患儿家属放弃治疗。文献复习中,糖皮质激素、IVIG和血浆置换均被作为一线治疗选择,大部分患儿都可以得到缓解甚至完全治愈。
儿童CIDP首次接受激素、IVIG或血浆置换治疗后初始有效率80%~100%[25];随访约70%可以完全缓解,部分遗留轻微运动障碍或疲劳[25]。本文病例初始治疗后93%(14/15)明显缓解,随访显示,33%(5/15)患儿完全缓解,60%(9/15)患儿仅遗留轻度疲劳或运动障碍。本文病例完全缓解比例低于既往报道,可能与本研究病例数较少、部分遗留有轻度疲劳或运动障碍的患儿仍在接受治疗和随访中有关。与成人相比,儿童CIDP预后较好,未达到完全缓解的患儿常仅遗留轻微运动障碍[26]。
1982年,Dyck等首次用慢性炎性脱髓鞘性多发性神经病命名并提出第1套临床诊断标准[27],要求病程进展>6个月。1991年美国神经病学会年会(ANN)制定的诊断标准[5]中,疾病进展时间>2个月。近20多年来,CIDP病例报道不断增加。有研究指出,16%的CIDP患者的起病后进展较快,在4~8周内达到疾病高峰[28]。2002年ENMC会议更新了儿童CIDP的诊断标准[2],患儿出现持续4周以上的进展性上肢或者下肢肌无力表现则考虑儿童CIDP。近年来报道的儿童CIDP病例[7]及本文病例中均有1/3左右的亚急性起病的患儿,在起病后4~8周内就达到的疾病高峰期。本文文献复习显示,亚急性起病的CIDP患儿,其感觉障碍和颅神经受累情况比慢性起病患儿多见,且更容易出现反复发作;通常首次发病时对于治疗药物的反应良好,随访中可出现多次反复发作。两组患儿在脑脊液异常、神经传导异常方面差异无统计学意义;治疗反应较好,预后较好,大多数可以正常进行日常生活,严重的后遗症较少。这与其他的小样本的相关研究中得出的结论相符合[7]。
综上所述,与成人CIDP相比,儿童CIDP较少见,亚急性起病较多,通过及时治疗可以有效缓解功能障碍。儿童CIDP亚急性起病患儿感觉障碍、颅神经异常多于慢性起病患儿,并且疾病发展过程中容易复发。
[1]EllrichmannG,GoldR,AyzenbergI,etal.Twoyears'long-termfollowupinchronicinflammatorydemyelinatingpolyradiculoneuropathy:efficacyofintravenousimmunoglobulintreatment.TherAdvNeurolDisord, 2017,10(2):91-101.
[2]NevoY,TopalogluH. 88thENMCinternationalworkshop:childhoodchronicinflammatorydemyelinatingpolyneuropathy(includingreviseddiagnosticcriteria),Naarden,TheNetherlands,December8-10, 2000.NeuromusculDisord, 2002,12(2):195-200
[3]中华医学会神经病学分会神经肌肉病学组,中华医学会神经病学分会肌电图及临床神经电生理学组,中华医学会神经病学分会神经免疫学组.中国慢性炎性脱髓鞘性多发性神经根神经病诊疗指南. 中华神经科杂志,2010,43(8):586-588
[4]QuinnTJ,DawsonJ,WaltersMR,etal.ReliabilityofthemodifiedRankinScale:asystematicreview.Stroke,2009,40(10):3393-3395
[5]NListed.Researchcriteriafordiagnosisofchronicinflammatorydemyelinatingpolyneuropathy(CIDP).ReportfromanAdHocSubcommitteeoftheAmericanAcademyofNeurologyAIDSTaskForce.Neurology,1991,41(5):617-618
[6]VandenBerghPY,HaddenRD,BoucheP,etal.EuropeanFederationofNeurologicalSocieties/PeripheralNerveSocietyGuidelineonmanagementofchronicinflammatorydemyelinatingpolyradiculoneuropathy:reportofajointtaskforceoftheEuropeanFederationofNeurologicalSocietiesandthePeripheralNerveSociety-firstrevision.EurJNeurol,2010,17(3):356-363
[7]CabassonS,TardieuM,MeunierA,etal.ChildhoodCIDP:Studyof31patientsandcomparisonbetweenslowandrapid-onsetgroups.BrainDev, 2015, 37(10):943-951
[8]RyanMM,Grattan-SmithPJ,ProcopisPG,etal.Childhoodchronicinflammatorydemyelinatingpolyneuropathy:clinicalcourseandlong-termoutcome.NeuromusculDisord,2000,10(6):398-406
[9]RossignolE,D'AnjouG,LapointeN,etal.Evolutionandtreatmentofchildhoodchronicinflammatorypolyneuropathy.PediatrNeurol, 2007,36(2):88-94
[10]LucchettaM,VidalE,SartoriS,etal.Long-termplasmaexchangeinpediatricCIDP.JClinApher, 2015,30(6):364-366
[11]JhaS,AnsariM,SonkarK,etal.Unusualfeaturesinchronicinflammatorydemyelinatingpolyneuropathy:Goodoutcomeafterprolongedventilatorysupport.JNeurosciRuralPract, 2011,2(2):171-173
[12]TaySY,ChanWP.A9-year-oldfemalewithbilaterallegweaknessafterinfluenzavaccination.PediatrAnn,2014,43(11):440-441
[13]RiekhoffAG,JadoulC,MercelisR,etal.Childhoodchronicinflammatorydemyelinatingpolyneuroradiculopathy—threecasesandareviewoftheliterature.EurJPaediatrNeurol,2012,16(4):315-331
[14]D'AmicoA,CatterucciaM,DeBenedettiF,etal.Rituximabinachildhood-onsetidiopathicrefractorychronicinflammatorydemyelinatingpolyneuropathy.EurJPaediatrNeurol,2012,16(3):301-303
[15]NirupamN,SharmaS,AnejaS,etal.Childhoodchronicinflammatorydemyelinatingpolyneuropathyassociatedwithacquiredscoliosis:acasereport.JChildNeurol,2012,27(6):804-806
[16]JoHY,ParkMG,KimDS,etal.Chronicinflammatorydemyelinatingpolyradiculoneuropathyinchildren:characterizedbysubacute,predominantlymotordominantpolyeuropathywithafavorableresponsetothetreatment.ActaNeurolScand,2010,121(5):342-347
[17]MarkowitzJA,JesteSS,KangPB.Childneurology:chronicinflammatorydemyelinatingpolyradiculoneuropathyinchildren.Neurology,2008,71(23):e74-e78
[18]RostasyKM,DiepoldK,BuckardJ,etal.Progressivemuscleweaknessafterhigh-dosesteroidsintwochildrenwithCIDP.PediatrNeurol,2003,29(3):236-238
[19]沈思翔, 冯建华, 周柏林, 等. 儿童慢性炎症性脱髓鞘性多发性神经病一例. 中华儿科杂志,2003,41(9):707-708
[20]HattoriN,IchimuraM,AokiS,etal.Clinicopathologicalfeaturesofchronicinflammatorydemyelinatingpolyradiculoneuropathyinchildhood.JNeurolSci,1998,154(1):66-71
[21]IijimaM,KoikeH,HattoriN,etal.PrevalenceandincidenceratesofchronicinflammatorydemyelinatingpolyneuropathyintheJapanesepopulation.JNeurolNeurosurgPsychiatry, 2008,79(9):1040-1043
[22]HafsteinsdottirB,OlafssonE.Incidenceandnaturalhistoryofidiopathicchronicinflammatorydemyelinatingpolyneuropathy:apopulation-basedstudyinIceland.EurNeurol,2016,75(5-6):263-268
[23]VandenBerghPY,RajaballyYA.Chronicinflammatorydemyelinatingpolyradiculoneuropathy.PresseMed,2013,42(6Pt2):e203-e215
[24]ChangSJ,LeeJH,KimSH,etal.Chronicinflammatorydemyelinatingpolyneuropathyinchildren:areportoffourpatientswithvariablerelapsingcourses.KoreanJPediatr,2015,58(5):194-198
[25]Al-BloushiMAS,HabeebYK,Al-JumahES.Chronicinflammatorydemyelinatingpolyradiculoneuropathyintwochildren.KuwaitMedJ, 2009,41(2):156-161
[26]DesaiJ,Ramos-PlattL,MitchellWG.Treatmentofpediatricchronicinflammatorydemyelinatingpolyneuropathy:Challenges,controversiesandquestions.AnnIndianAcadNeurol,2015,18(3):327-330
[27]DyckPJ,O’BrienPC,OviattKF,etal.Prednisoneimproveschronicinflammatorydemyelinatingpolyradiculoneuropathymorethannotreatment.AnnNeurol, 1982, 11(2): 136-141
[28]AnadaniM,KatirjiB.Acute-onsetchronicinflammatorydemyelinatingpolyneuropathy:Anelectrodiagnosticstudy.MuscleNerve,2015,52(5):900-905
(本文编辑:张崇凡,孙晋枫)
Clinicalcharacteristicsof15caseswithchildhoodchronicinflammatorydemyelinatingpolyneuropathyandliteraturereview
QIUTian,CHAIYi-ming
(DepartmentofNeurology,Children'sHospitalofFudanUniversity,Shanghai201102,China)
Corresponding Author: Chai Yi-ming, E-mail: acyimm@hotmail.com
ObjectiveTo investigate the clinical characteristics and prognosis of chronic inflammatory demyelinating polyneuropathy (CIDP) in children. MethodsClinical data were collected and analyzed from patients with CIDP who visited the Department of Neurology, Children's Hospital of Fudan University between January 1,2008 and January 31,2017. PubMed databases were retrieved from January 1991 to February 2017 with CIDP cases in children. According to the peak time of the disease(4-8 weeks for subacute onset group, >8weeks for chronic onset group), the retrieved literature cases and the cases in the study were aggregated and divided into two groups, They were compared in clinical manifestation, laboratory examination, treatment and prognosis. ResultsFirstly, there were 15 CIDP cases who met the criteria of inclusion and exclusion, including 6 males and 9 females; 4 cases between 1-3 years old, 4 cases between 4-7 years old, 7 cases between 8-15 years old; the age of disease onset was 1-11 years old;7 patients had a history of prodromal infection; development to the peak of the disease required 4 weeks to 13 months, with an average (12.93±13.57) weeks. At the beginning of the disease all cases showed dyskinesia;4 cases with sensory disorder; the peak of the disease MRS score: 3 points in 8 cases, 4 points in 6 cases and 5 points in 1 case; the course of disease was monophase in 3 cases, progressive in 5 cases and recurrent in 7 cases;all cases had nerve electrophysiological changes; in all cases dissociation of protein from cell in CSF occurred. 14 cases were treated with glucocorticoid, 1 case was treated with intravenous immunoglobin; 1 case was discharged from hospital voluntarily and 14 cases were hospitalized between 7-17 days with the clinical symptoms improved;during the follow-up period, 7 cases were relapsed.Secondly, 87 cases were retrieved from PubMed, together with our 15 cases,102 cases were analyzed. There were 38 patients with subacute-onset disease and 64 patients with chronic-onset disease. Patients exhibited symptoms in less than 2 months with more often sensory abnormalities (57.1%vs23.5%,P=0.002), and cranial nerve abnormalities (25.7%vs7.8%,P=0.023). There was no significant difference between the two groups in gender, age of disease onset and movement disorders. Patients with subacute-onset relapsed more than patients with chronic-onset(62.2%vs34.0%,P=0.010).There were more monophasic evolution in chronic-onset disease than in subacute-onset disease (50.8%vs27.8%,P=0.026).ConclusionChildren with subacute onset of CIDP are slightly more than adults, and their sensory abnormalities and cranial nerve abnormalities are more frequent than children with chronic onset of CIDP, and they are prone to relapse during disease development. Dyskinesia is common in children with CIDP, and cranial neuropathy and respiratory failure are unusual. Nerve electrophysiological changes and dissociation of protein from cell in CSF are common in CIDP., and early diagnosis and initial treatment are important.
Chronic inflammatory demyelinating polyradiculoneuropathy; Clinical analysis; Children
复旦大学附属儿科医院神经科 上海,201102
柴毅明,E-mail: acyimm@hotmail.com
10.3969/j.issn.1673-5501.2017.03.06
2017-05-16
2017-06-08)
