肌萎缩侧索硬化合并垂体腺瘤1例并文献复习
2017-06-29袁小闯杨帅凯王炳浩苏魏巍杨伟民滕军放
袁小闯 韦 星 杨帅凯 王炳浩 苏魏巍 杨伟民 滕军放
郑州大学第一附属医院神经内科 郑州 450052
肌萎缩侧索硬化合并垂体腺瘤1例并文献复习
袁小闯 韦 星 杨帅凯 王炳浩 苏魏巍 杨伟民 滕军放△
郑州大学第一附属医院神经内科 郑州 450052
目的 通过1例肌萎缩侧索硬化合并垂体腺瘤病例报道并文献复习,探讨其发病的病理机制,以便找到针对病因的治疗方法。方法 分析我院收治的1例肌萎缩侧索硬化合并垂体腺瘤病例资料并查阅国内外文献。 结果 肌萎缩侧索硬化发病机制可能与胰岛素样生长因子1有关。 结论 肌萎缩侧索硬化的发病机制可能与胰岛素样生长因子1有关,具体发病机制需要进一步的研究。
肌萎缩侧索硬化;垂体腺瘤;肢端肥大症;胰岛素样生长因子1
肌萎缩侧索硬化是严重的神经系统变性疾病,具有严重的致残和致命性。主要累及脊髓前角细胞、脑干运动神经核团及椎体束。国外报道的一些罕见病例中,高胰岛素血症、甲状腺功能亢进及甲状旁腺功能亢进伴随着一些肌萎缩侧索硬化相似的症状和体征。 但肌萎缩侧索硬化合并垂体腺瘤国内外报道很少,研究其相关机制不是十分明确。现报道我院收治的1例肌萎缩侧索硬化合并垂体腺瘤的患者,归纳其行垂体腺瘤术及口服溴隐亭药物治疗前后的临床变化,分析其潜在病理机制,提高对疾病的认识。
1 临床资料
患者男性,42岁,主因“四肢无力6个月余”于2015-12-12收住我院神经内科。患者6月余前无明显诱因出现双下肢无力、僵硬,上下楼梯时明显,生活可基本自理。4个月前患者自觉左上肢无力,双下肢无力较前加重,并自觉有“肉跳感”,至当地医院就诊,行头颈部磁共振提示“头部磁共振未见异常,颈椎间盘突出”。考虑“颈椎病”,给予营养神经药物治疗,症状未见减轻。2个月前患者可上4层楼梯,近2周患者仅可上2层楼梯即感觉肌肉无力、酸痛。患者自发病来以来,偶有肢体近端肉跳感,肢体力弱无晨轻幕重现象,无明显肢体麻木、胸腹部束带感,无吞咽困难及尿便障碍。患者出生发育快于健康同龄儿,既往高血压2 a,未规律服用降压药物;糖尿病4月,未正规治疗。父母非近亲结婚,有1姐1弟1妹2子均体健。入院体检:血压 147/95 mmHg(1 mmHg=0.133 kPa),肢端肥大症体貌,心肺腹检查未见明显异常。意识清楚,语言流利,高级皮质功能正常。眼睑无下垂,眼球运动充分,双侧鼻唇沟对称,舌肌未见萎缩,余脑神经检查未见明显异常。双上臂及双手掌大小鱼际肌萎缩,左上肢肌力Ⅳ级,右上肢肌力Ⅴ级,双下肢肌力Ⅳ级,四肢肌张力痉挛性增高,双上肢腱反射增强(+++),双下肢腱反射亢进(++++),踝阵挛阳性,双侧巴宾斯基征阳性。四肢深浅感觉检查未见明显异常。共济失调检查未见明显异常。入院生化检查示:血常规、肝肾功能、红细胞沉降率、C反应蛋白、甲状腺功能及抗体、抗核抗体均未见明显异常。胰岛素样生长因子1:948 ng/mL↑(101~267 ng/mL),促黄体生成素、促卵泡激素、雌二醇未见异常,睾酮:1.58 ng/mL↓(1.75~7.81 ng/mL),泌乳素:18.97 ng/mL↑(2.64~13.13 ng/mL)。肌电图示广泛神经源性损害,可见束颤电位和正锐波,病变涉及颈胸腰骶段运动神经。颈髓磁共振未见明显异常,头部垂体薄层扫描示鞍区占位性病变,考虑垂体瘤。头DTI示双侧皮质脊髓束、双侧内侧丘系延髓段稀疏,额叶层面少量纤维束中断。见图1。依据E1 Escorial 会议制定的ALS诊断标准及Airlie House ALS诊断标准,诊断为:(1)肌萎缩侧索硬化;(2)垂体瘤;(3)高血压1级低危;(4)Ⅱ型糖尿病。患者于2016-01行垂体瘤γ刀手术及口服溴隐亭治疗。2016-05因肢体无力症状加重再次我科住院,检查:血压 130/80 mmHg,肢端肥大症体貌,心肺腹检查未见明显异常。意识清楚,语言流利,高级皮质功能正常。眼睑无下垂,眼球运动充分,双侧鼻唇沟对称,舌肌未见萎缩,余脑神经检查未见明显异常。双上臂及双手掌大小鱼际肌萎缩,左上肢肌力Ⅳ-级,右上肢肌力Ⅳ级,双下肢肌力Ⅳ-级,四肢肌张力减低,双上肢腱反射增强(+++),双下肢腱反射亢进(++++),踝阵挛阳性,双侧巴宾斯基征阳性。四肢深浅感觉检查未见明显异常。共济失调检查未见明显异常。复查肌电图仍符合广泛神经元性损害。
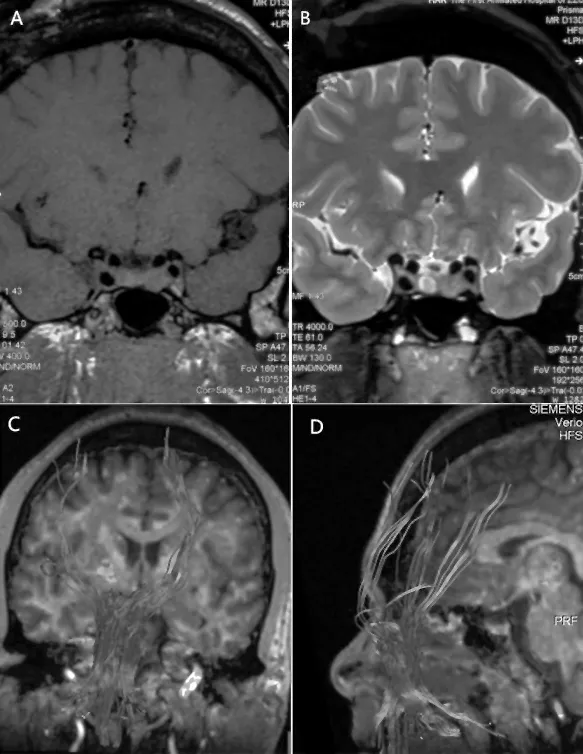
图1 A:垂体占位磁共振T1低信号; B:垂体占位磁共振T2高信号; C、D头部DTI示皮质脊髓束、皮质脑干束稀疏、部分中断
2 讨论
肌萎缩侧索硬化是运动神经元病中最常见的一种类型,于1830年由Bell首次对该病患者临床表现做过描述,1896年法国神经病学家Charcot对该病的临床及病理做了明晰的阐述。该病病程短(3~5 a),致残致死率高,60%在起病3 a内死亡[1]。本病主要累及脊髓前角细胞、脑干运动神经核团及椎体束。
病因及发病机制不明,暴露于毒素和重金属、病毒感染、DNA修复机制缺陷、免疫系统功能紊乱、代谢异常和内分泌功能紊乱是潜在因素。国外文献统计,5%~10%的肌萎缩侧索硬化患者具有家族遗传性,成人型主要为常染色体显性遗传,青年型为常染色体显性或隐性遗传。约25%突变致病基因中为编码铜锌超氧化物歧化酶的基因,定位于21号染色体长臂21q22.1-22.2,目前已发现至少160种突变,近年有证据显示基因突变产生毒性蛋白从多方面影响运动神经元功能[2]。目前发现一种青少年起病的常染色体隐性遗传家族性肌萎缩侧索硬化患者是由编码alsin蛋白的基因突变导致。发现的另一种常染色体隐性遗传家族性肌萎缩侧索硬化与senataxin基因突变有关。然而,在大多数散发性的肌萎缩侧索硬化患者,未发现与遗传因素有十分明确的关系。有证据表明,肌萎缩侧索硬化发病可能有免疫机制参与,外周血及脑脊液可以检测出抗神经元结构组分抗体,脑脊液中抗体水平高于血清,用患者血清对神经元培养有毒害作用。尸检发现肌萎缩侧索硬化患者的脊髓及大脑运动皮质有大量的星型细胞或小胶质细胞,血管周围和运动前脚可见T淋巴细胞浸润。也有报告表明,肌萎缩侧索硬化与中毒因素有关,曾有报告表明重金属中毒患者并发有脊髓性和根性运动症状的报告。但无直接证据表明重金属中毒可直接导致本病。肌萎缩侧索硬化的主要病理表现为脑干运动神经核团的神经元细胞及脊髓前角细胞缺失,颈椎的大运动神经元细胞变性早期显著出现。运动神经元显示轴突变性、脱髓鞘、轴突侧支形成,大的髓鞘纤维不成比例的缺失。
肌萎缩侧索硬化大部分在50岁后起病,30岁前发病较少见,男性发病率高于女性一倍,起病较为隐匿,进展缓慢,早期以远端肢体无力起病,手指活动无力笨拙,不能完成精细动作,之后发现手部肌肉萎缩,如大小鱼际肌、蚓状肌、骨间肌等萎缩,并逐渐向前臂、上臂、肩部肌群发展,表现为无力、萎缩及肌束颤动。体征早期表现为肌无力和肌萎缩,远端重于近端,腱反射可异常亢进,病理征阳性,上肢腱反射减弱或消失提示颈膨大前脚细胞严重受损。括约肌障碍比较少见,可出现呼吸机受累,长期卧床,呼吸困难,咳嗽无力,死于肺部感染。有些病例可合并痴呆出现[3],但无意识障碍。具有家族遗传性的患者起病早,生存期短。
肌萎缩侧索硬化的诊断必须具有临床、电生理或神经病理下运动神经元损害的证据,排除可以解释临床和电生理特征的其他疾病。主要与颈椎型颈椎病、多灶性运动神经元病、平山病、脊髓型肌萎缩症、脊髓空洞症、良性肌束震颤、多发性硬化、包涵体肌炎、脊髓肿瘤等鉴别诊断。临床确诊的肌萎缩侧索硬化要确定脑、颈、胸、腰骶神经支配区中的3个区域存在上下运动神经元损害临床证据。肌电图表现为肌肉放松时的广泛纤颤电位及正锐波,以及束颤电位和主动收缩时的电位时限增宽、波幅增大。
该例患者肌肉萎缩,肌张力增高,腱反射亢进,锥体束征阳性,肌电图示广泛神经源性损害,头部DTI成像示皮质脊髓束稀疏,符合原始和修订EL Escoliar ciriteria 临床确诊肌萎缩性侧索硬化症的标准。患者头部垂体高分辨率磁共振示垂体占位、垂体腺瘤,及患者有肢端肥大症的体征,符合垂体腺瘤的诊断。本例病例是ALS患者合并垂体瘤,在药物溴隐亭及放射治疗垂体瘤后,患者肢体的肌张力增高的症状有所缓解,肌无力及肌萎缩症状较治疗前加重。其病理机制可能与垂体分泌的某种物质有关。查阅国内外的文献,报道ALS合并内分泌疾病的病例不是很多。国外报道的一些罕见病例中,高胰岛素血症、甲状腺功能亢进、甲状旁腺功能亢进伴随着一些ALS相似的症状。此外,有很多研究者注意到ALS患者中胰岛功能存在障碍。2007年雅典Evangelia Kararizou报道1例诊断为ALS的患者合并有垂体腺瘤的患者在接受溴隐亭治疗3个月后患者的ALS症状有所缓解[4]。也有一些患者并发恶性肿瘤,在肿瘤好转时ALS的症状亦有缓解。2010年Mondok等[5]报道1例垂体微腺瘤合并ALS的患者在垂体瘤切除术后病情快速进展,最后死于呼吸衰竭。推测病情的进展加速与胰岛素样生长因子1有关。胰岛素样生长因子1是一种可作用于多种组织和器官的多向蛋白,可提高神经细胞生长存活率,介导神经细胞生长,已应用于生长发育不良、骨质疏松症、糖尿病等的治疗。目前对于胰岛素样生长因子1和ALS的关系研究还不是十分明确。缑元冲等[6]认为胰岛素样生长因子1治疗ALS的体外实验和体内实验效果显著。近年来有些研究认为血清和脑脊液中的胰岛素样生长因子1水平不成线性先关,考虑与血脑屏障有关系。Bilic等[7]研究35例运动神经元病患者和40例健康者比较脑脊液中胰岛素样生长因子1的含量,发现患有运动神经元者胰岛素样生长因子1含量明显减少。胰岛素样生长因子1治疗ALS的效果临床试验做的较少,缺少大规模的多中心随机对照研究。应当加强对ALS发病机制的研究,以便找到新的药物作用靶点,改善疾病的预后。
[1] 杨琼,樊东升.肌萎缩侧索硬化症临床诊断进展[J].中国现代神经疾病杂志,2012,6(12):245-251.
[2] 张华纲,唐璐,张楠,等.中国家族性肌萎缩侧索硬化患者SOD1基因突变与临床表型研究[J].中华神经科杂志,2012,45(7):453-458.
[3] 樊东升.重视ALS患者的认知和行为损害[J].中华神经科杂志,2010,43(9):603-606.
[4] Evangelia Kararizou,Elevtherios Stamboulis,Ioannis Ma-rkou,et al.Amyotrophic lateral sclerosis and prolactinoma[J].Functional Neurology,2007,22(1):39-41.
[5] Mondok A1,Aranyi Z,Kovacs GG,et al.Rapid progression of amyotrophic lateral sclerosis in an acromegalic patient after surgical resection of a growth hormone-producing pituitary adenoma.[J].Neurologist,2010,16(5):315-318.
[6] 缑元冲,李春岩.胰岛素样生长因子-1 系统和肌萎缩侧索硬化[J].脑与神经疾病杂志,2006,14(6):468-469.
[7] Bilic E,Bilie E,Rudan I,et al.Comparison of the growth hormone,IGF-I and insulin in cerebrospinal fluid and serum between patients with motor neuron disease and healthy controls[J].Eur J Neuro,2006,13(12):13 400.
(收稿2016-11-02)
R736.4
D
1673-5110(2017)05-0142-03
△通讯作者:滕军放,男,主任医师、教授、博士生导师,郑州大学第一附属医院内二医学部主任,神经内科二病区主任,E-mail:13838210077@163.com
