低钾型周期性麻痹与CACNA1S基因相关的研究进展
2017-03-13何妍妍陈勇军
何妍妍,邹 伟,陈勇军
(南华大学附属南华医院神经内科,湖南衡阳421002)
低钾型周期性麻痹与CACNA1S基因相关的研究进展
何妍妍,邹 伟,陈勇军
(南华大学附属南华医院神经内科,湖南衡阳421002)
近年来随着分子生物学、遗传学的进展,越来越多的证据表明低钾性周期性麻痹是一种与离子通道基因相关的遗传性疾病,目前发现的突变基因有CACNA1S、SCN4A、KCNE3,其中以CACNA1S所发现的突变率最高,我们拟对近年以来发现的CACNA1S基因突变及相关功能研究做一综述。
低钾型周期性麻痹;遗传性疾病,先天性;基因
低钾型周期性麻痹(hypokalemic periodic paralysis,HOKPP)是一种常染色体显性疾病,可分为原发性低钾型周期性麻痹(primary hypokalemic periodic paralysis,PHOKPP)及继发性低钾型周期性麻痹。而PHOKPP中又包括家族性低钾型周期性麻痹(familial hypokalemic periodic paralysis,FHOKPP)以及散发性低钾型周期性麻痹(sporadical hypokalemic periodic paralysis,SHOKPP),有部分学者甚至把Andersen-Tawil综合征也归纳为原发性周期性麻痹中的一种,我国患者发病以SHOKPP多见,西方国家则以FHOKPP多见[1]。继发性低钾型周期性麻痹病因包括甲状腺功能亢进、肾小管酸中毒、Ig A肾病、醛固酮增多症等,在亚洲,尤其是中国及日本,最主要的类型为继发于甲状腺功能亢进的甲亢性低钾型周期性麻痹。
近年来随着分子生物学、遗传学的进展,越来越多的证据表明低钾型周期性麻痹是一种与离子通道基因相关的常染体显性遗传性疾病,目前发现的突变基因有CACNA1S、SCN4A、KCNE3,其中以CACNA1S所发现的突变率最高,西方国家统计约占患病人数的69%[2],CACNA1S基因上目前发现的突变位点包括:R528 H/G、876E、R897S、R900G/S、H916Q、R1239H/G。Sternberg分析数据后指出CACNA1S突变中以R528G、R1239 H最常见[2]。本文拟对近年以来发现的CACNA1S基因突变及相关功能研究做一综述。CACNAIS相关突变位占见表1。
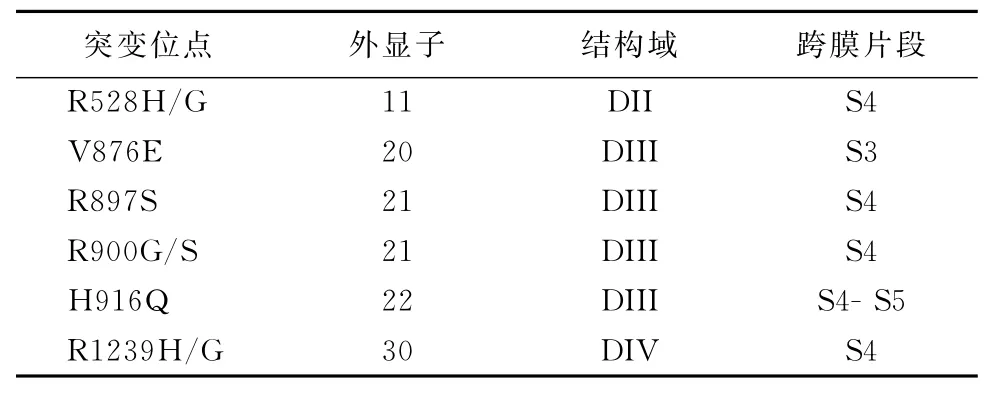
表1 CACNA1S基因上的突变位点
1 HOKPP的CACNA1S基因相关突变
1.1 HOKPP与R528 H R528H突变较常见,Sternberg等[2]调查了HOKPP患者58例,其中40例患者与突变的CACNA1S相关,证实发生R528 H突变26例,突变率为45.0%;Miller等[3]调查了HOKPP患者71例,42例CACNA1S突变的患者中有17例发生R528 H突变,突变率为40.5%。R528 H突变所致的低钾型周期性麻痹的临床特点是发病年龄较早、低钾情况不重,症状较轻,不引起呼吸困难,可有肌肉疼痛症状。Kawamura等[4]指出R528H突变的患者首次发病时间为(14±3)岁。在发作时血钾浓度方面,Miller等[3]在2004年对HOKPP患者进行数据分析后指出,HOKPP发作时血钾浓度(2.9 ±0.7)mmol/L,但Sternberg等[2]研究样本后总结的发作时血钾浓度较低,为(1.69±0.49)mmol/L,这可能与样本量小有关,需扩大样本量继续验证。研究发现R528 H有不完全外显率,突变的女性携带者可以无临床症状或轻的表型,但是这种突变在子代和男性的外显率高、表现严重[5]。动物实验也支持这一临床现象,雄性R528H小鼠比具有相同突变的雌性小鼠有更严重的表现[6],目前还不明确是否与雌激素有关。Sternberg等[2]对大量的HOKPP患者的组织病理进行了研究发现R528H突变患者肌肉活检表现为单纯的肌纤维空泡样变病理改变。治疗上可予以乙酰唑胺降低发作频率,也有报道称乙酰唑胺治疗无明显效果,甚至可以加重病情[7]。有学者提出可予以氯离子阻断剂布美它尼预防再发,其机制为布美他尼可抑制异常去极化及细胞兴奋性的丧失从而治疗低钾型周期性麻痹,现阶段的研究表明在小鼠及离体细胞中治疗是有效的,尚未在人类中进行该项药物实验[8]。
1.2 HOKPP与R528G Wang等[9]于2005年首次报道了我国的1个R528G突变的家系,该家系的临床特点为患者的发病频率与年龄的关系曲线呈倒U型,发病频率及发病年龄与性别无关,女性存在不完全外显率。家系成员的发病年龄为3~23岁,40岁以后发病频率减少,60岁之后消失。女性外显率为83.3%(10/12),男性外显率为100%(9/9)。临床症状为以双下肢乏力为主的四肢乏力,未报导呼吸困难,但Kil等[10]在2009年报道了1例16岁R528G突变的男性患者,他的发病特点主要表现为四肢和躯干的严重迟缓性麻痹及呼吸窘迫,当时测得血钾2.7mmol/L,补钾后症状缓解。口服乙酰唑胺对病情预防无效,予以螺内酯、阿米洛利及钾盐联合治疗后发作次数减少。
1.3 HOKPP与V876E V876E突变首次由Ke等[11]于2009年研究一南美HOKPP家系时发现,此突变的临床特点为发病年龄早、完全外显、预后严重,男性患者临床表现较女性患者严重,家族中甚至有2例男性患者出现呼吸抑制而死亡。我国于2015年报告了1例散发的V876E突变的女性低钾麻痹患者,发作时有心悸、呼吸急促[12]。V876E突变发病的平均年龄为(5.2±3.6)年,比R528H突变及R1239H突变均早,南美发病家系中甚至有一个出生后1年内开始发生症状[11]。较R528 H及R1239 H突变不同,V867E基因突变的男女携带者均患病,表现为完全外显。治疗上钾剂治疗效果较乙酰唑胺好。
1.4 HOKPP与R897S、H916Q R897S、H916Q均较少见。目前仅发现1例具有土耳其血统的男性患者被报道存在R897S突变,患者发病年龄早,出生时即有呼吸窘迫、肌张力减退,但当时血钾正常,患者在1岁时,观察到有短暂性行走困难和瘫痪现象,多在夜晚发作,每次持续时间10~30分钟,血钾最低可达2.4 mmol/L。患者随着年龄增长,发作频率以及严重程度均减轻,但持续时间延长,可达数小时,治疗上予以钾盐及乙酰唑胺均有效[13]。H916Q也仅见我国于2012年报道的1个家系[14],其特点如下:首发年龄(19±1)岁,发作频率1~7次/年,以清晨及夜间发病为主,每次发病持续时间2~12小时,存在不完全外显率,所有男性携带者(10例)均患病,暂未发现女性携带者(4例)有明显的临床表现。
1.5 HOKPP与R900G、R900S R900G、R900S在同一位点被不同的氨基酸取代,临床特点表现不同。Hirano等[15]于2011年报道发现1个R900G突变的家系,此突变的特征是:寒冷不诱发低钾麻痹的症状、发病后有血钾水平升高甚至出现高钾血症的现象、钾剂及乙酰唑胺治疗有效。Matthews等[16]于2009年首次发现R900S突变,但未描述其临床特点,Ke等[17]在2015年报道了一个R900S突变所致的低钾麻痹的家系,患者可被寒冷诱发低钾麻痹,对乙酰唑胺治疗无效,但联合氨苯蝶啶及氯化钾治疗后发作频率可减少,有不完全外显率,所有的男性突变携带者(3例)均发病,所有3个女性突变携带者(3例)无症状。
1.6 HOKPP与R1239 H R1239 H也较为常见,Sternberg等[2]分析样本后指出R1239 H突变占24.0%。和R528 H突变相似,症状的严重程度与年龄的曲线呈倒U型关系,患者儿童期即可发病,青春期病情逐渐加重,大多数患者30岁后症状的严重程度开始减轻、发作频次开始减少,甚至可以不再发病[18]。有学者认为R1239H发病年龄较R528H早、发作时血钾水平较R528 H低[19]。Kawamura等[4]指出R1239 H的首次发病时间是(10±5)岁,比R528 H早(7±4)岁。在发病时血钾浓度方面,Miller等[3]分析样本后得出发作时血钾浓度为(1.9±0.4)mmol/L,而Sternberg等[2]指出R1239 H发作时血钾浓度为(2.23±0.86)mmol/L,较R528H高。Kawamura与Sternberg分析样本后得出的R1239H与R528H发作时血钾浓度的差异性结果完全相反,因此还需继续扩大样本量明确不同的突变类型间发作时血钾浓度有无差异。R1239 H也存在不完全外显率,女性携带者可无症状或症状轻微。予以乙酰唑胺治疗可降低发作频率,但也有报道称引起肌无力加重[7]。2014年有学者使用托吡酯成功降低了一例反复发作、使用乙酰唑胺治疗效果欠佳的R1239H突变低钾麻痹型患者的发病次数及严重程度[20],提示托吡酯对治疗低钾型周期性麻痹有效,但其作用机制尚未完全明确。
1.7 HOKPP与R1239G 韩国学者Kim等[21]于2005年报道了1个R1239G突变的家系,患者首次发病较早,在3~14岁之间,每天可多次发作,一般在晚上或早上发病,下午恢复正常,女性月经前期发作频率增加,呼吸抑制、心律失常少见。随病情发展,发作频率及程度都会进一步加快或加重;男性患者病情的严重程度随着年龄的增长而加重,但女性患者的发病频率以及严重程度却随着年龄的增长而减轻。男女性患者均对钾剂和乙酰唑胺无效,甚至使用乙酰唑胺有病情加重的情况,而安体舒通、螺内酯治疗后症状明显改善。Winczewska-Wiktor等[22]也在2007年对波兰存在这一突变的病例进行了报导,临床特点基本与韩国报道相似。
2 CACNA1S突变导致HOKPP机制
CACNA1S基因位于染色体1q31-32上,编码L-型钙通道的α1亚基,L-型钙通道由α1、α2、β、γ和δ5个亚基组成,其中α1亚基是离子通道的主要的功能单位[23],其他亚基起辅助调节作用。α1亚基包含有(DI-DIV4)4个同源结构域,它们对称排列,中间围成亲水通道,每一个结构域还包括(S1-S6)6个跨膜螺旋结构片段,其中S1-S4是电压传感区[11],而S5和S6是孔道区,S4片段含有带正电荷的氨基酸,随膜电位的变化而发生构象变化,调节通道的开闭,是电压感受器的关键。目前发现CACNA1S突变主要影响S4片段,以Ⅱ和Ⅳ的S4最多,仅有V876E影响了S3片段。CACNA1S如何导致低钾血症尚不清楚,有学者提出了以下假说:①门控电流假说:突变的S4片段因结构功能改变,形成一个独立于正常离子通道孔的附属离子通道,产生门控孔电流(Ⅰgp),Ⅰgp在静息电位时激活,去极化时关闭,静息电位时Ⅰgp因内向的质子或离子流导致细胞膜的去极化及Na+超载[11,23],当细胞外血钾水平正常时,去极化并不明显,但是当细胞外血钾水平降至3 mmol/L以下时,静息电位肌纤维去极化,产生电压依赖性离子通道失活、弛缓性肌肉麻痹、细胞内钠离子超载、肌细胞水肿等一系列低钾型周期性麻痹的特征[1,23-26],实验证明在CACNA1S突变的大鼠模型和患者肌纤维中可以检测到Ⅰgp电流[1,6,23]。而突变所致的异常Na+的内流,导致细胞内Na+浓度的升高,这可以刺激Na+-K+-ATP将细胞内多余的Na+移出细胞,同时把细胞外的K+移入细胞内,产生低钾血症,很多学者甚至认为Na+泵活性增强是HOKPP发生的最终通路[24,27]。但是患者平时不发病,可能因为机体的负反馈机制使机体内环境平衡,当诱因出现时,通过一系列级联放大反应,改变了Na+-K+-ATP与Na+的亲和力和最大反应速度,在突变所致的细胞内Na+浓度异常升高的情况下,改变调定点,使血钾降低,从而导致低钾型周期性麻痹。②钙通道假说:CACNA1S突变可直接影响L-型钙通道的α1亚基,α1亚基有骨骼肌双氢吡啶受体,而骨骼肌双氢吡啶受体可以激活钙通道使细胞内钙离子释放,从而引起骨骼肌收缩,S 4片段突变后钙离子通道受到影响,出现开放异常,导致Ca2+释放减少,这使得骨骼肌细胞膜的静息电位稳态以及阈值发生改变,不仅直接影响骨骼肌细胞的收缩,还可以直接或间接使钠通道失活,从而导致细胞膜不能兴奋性或兴奋性下降,出现肌无力症状[28]。
在治疗方面,首先是避免寒冷、输入加胰岛素的葡萄糖等诱因,其次是药物治疗,较常见的是钾盐及碳酸酐酶抑制剂,而碳酸酐酶抑制剂中又以乙酰唑胺使用最多,现也有学者将二氯苯磺胺用于临床,但目前缺乏大样本的临床资料[29]。碳酸酐酶抑制剂的治疗机制考虑与改变血液PH值相关,在周期型低钾性麻痹患者中,往往碳酸酐酶抑制剂对R被H替换者比对R被G替换者更有效,目前原因尚不明确,可能与R被H替换相较于R被G替换对血液PH值影响更大有关[7]。对于使用碳酸酐酶抑制剂无效的患者,还可使用保钾利尿剂,如氨苯蝶啶、阿米洛利等。随着医学技术的发展,在治疗方面也有新的进展,Wu等[8]于2013年提出可使用氯离子阻断剂布美他尼阻止氯离子内流从而抑制异常去极化及细胞奋性的丧失来治疗低钾型周期性麻痹,但该药尚无临床实验。2014年García-Sobrino等[20]使用托吡酯成功治疗了一例对乙酰唑胺治疗无效的患者,托吡酯也是通过影响PH值起效,但治疗效果与碳酸酐酶抑制剂不同,暂不排除托吡酯有影响Na+通道或其他作用机制。
综上所述,HOKPP是一类与基因突变相关的遗传性疾病,突变位点的不同及替代的氨基酸的不同临床表现存在差异,对碳酸酐酶抑制剂的治疗效果也不同。目前发现CACNA1S突变所占比例最大,其中又以R528H、R1239H较常见,主要影响Ⅱ、Ⅳ的S4片段,仅有V876E影响了S3片段,暂未发现有突变影响其他片段。基因突变点的确定对疾病诊断及基因治疗甚至产前筛查均有重要意义,目前我们对于HOKPP的少见类型及临床病例研究资料仍不足,是否存在尚未发现的突变影响了S1、S2、S6片段仍不清,还需进一步扩大样本研究。
[1]Tricarico D,Camerino DC.Recent advances in the pathogenesis and drug action in periodic paralyses and related channelopathies[J].Front Pharmacol,2011,2:8.
[2]Sternberg D,Maisonobe T,Jurkat-Rott K,et al.Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A[J].Brain,2001,124(Pt 6):1091-1099.
[3]Miller TM,Dias dSMR,Miller HA,et al.Correlating phenotype and genotype in the periodic paralyses[J].Neurology,2004,63(9):1647-1655.
[4]Kawamura S,Ikeda Y,Tomita K,Watanabe N,Seki K.A family of hypokalemic periodic paralysis with CACNA1S gene mutation showing incomplete penetrance in women[J].Intern Med,2004,43(3):218-222.
[5]Ikeda Y,Watanabe M,Shoji M.Mutation analysis of the CACNL1A3 gene in Japanese hypokalemic periodic paralysis families[J].Nihon Rinsho,1997,55(12):3247-3252.
[6]Wu F,Mi W,Hernández-Ochoa EO,et al.A calcium channel mutant mouse model of hypokalemic periodic paralysis[J].J Clin Invest,2012,122(12):4580-4591.
[7]Matthews E,Portaro S,Ke Q,et al.Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype[J].Neurology,2011,77(22):1960-1964.
[8]Wu F,Mi W,Cannon SC.Beneficial effects of bumetanide in a CaV1.1-R528H mouse model of hypokalaemic periodic paralysis[J].Brain,2013,136(Pt 12):3766-3774.
[9]Wang Q,Liu M,Xu C,et al.Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a Chinese family[J].J Mol Med(Berl),2005,83(3):203-208.
[10]Kil TH,Kim JB.Severe respiratory phenotype caused by a de novo Arg528Gly mutation in the CACNA1S gene in a patient with hypokalemic periodic paralysis[J].Eur J Paediatr Neurol,2010,14(3):278-281.
[11]Ke T,Gomez CR,Mateus HE,Castano JA,Wang QK.Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a South American family[J].J Hum Genet,2009,54(11):660-664.
[12]Yang H,Zhang H,Xing X.V876E mutation in CACNA1S gene associated with severe hypokalemic periodic paralysis in a Chinese woman[J].J Formos Med Assoc,2015,114(4):377-378.
[13]Chabrier S,Monnier N,Lunardi J.Early onset of hypokalaemic periodic paralysis caused by a novel mutation of the CACNA1S gene[J].J Med Genet,2008,45(10):686-688.
[14]Li FF,Li QQ,Tan ZX,et al.A novel mutation in CACNA1S gene associated with hypokalemic periodic paralysis which has a gender difference in the penetrance[J].J Mol Neurosci,2012,46(2):378-383.
[15]Hirano M,Kokunai Y,Nagai A,et al.A novel mutation in the calcium channel gene in a family with hypokalemic periodic paralysis[J].J Neurol Sci,2011,309(1-2):9-11.
[16]Matthews E,Labrum R,Sweeney MG,et al.Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis[J].Neurology,2009,72(18):1544-1547.
[17]Ke Q,He F,Lu L,et al.The R900S mutation in CACNA1S associated with hypokalemic periodic paralysis[J].Neuromuscul Disord,2015,25(12):955-958.
[18]柯青,吴卫平,徐全刚,等.家族性低钾型周期性麻痹的基因突变与临床特征[J].中华神经科杂志,2006,39(5):323-327.
[19]武昆.低钾型周期性麻痹家系基因CACNA1S和SCN4A突变分析[D].安徽:安徽医科大学,2013.
[20]García-Sobrino T,Pardo J.Successful treatment of hypokalemic periodic paralysis with topiramate[J].Eur J Neurol,2014,21(9):e73-74.
[21]Kim JB,Lee KY,Hur JK.A Korean family of hypokalemic periodic paralysis with mutation in a voltage-gated calcium channel(R1239G)[J].J Korean Med Sci,2005,20(1):162-165.
[22]Winczewska-Wiktor A,Steinborn B,Lehman-Horn F,et al.Myopathy as the first symptom of hypokalemic periodic paralysis--case report of a girl from a Polish family with CACNA1S(R1239G)mutation[J].Adv Med Sci,2007,52(Suppl 1):155-157.
[23]Jurkat-Rott K,Weber MA,Fauler M,et al.K+-dependent paradoxical membrane depolarization and Na+overload,major and reversible contributors to weakness by ion channel leaks[J].Proc Natl Acad Sci USA,2009,106(10):4036-4041.
[24]Kung AW.Clinical review:thyrotoxic periodic paralysis:a diagnostic challenge[J].J Clin Endocrinol Metab,2006,91(7):2490-2495.
[25]Sokolov S,Scheuer T,Catterall WA.Gating pore current in an inherited ion channelopathy[J].Nature,2007,446(7131):76-78.
[26]Francis DG,Rybalchenko V,Struyk A,et al.Leaky sodium channels from voltage sensor mutations in periodic paralysis,but not paramyotonia[J].Neurology,2011,76(19):1635-1641.
[27]Hsieh MJ,Lyu RK,Chang WN,et al.Hypokalemic thyrotoxic periodic paralysis:clinical characteristics and predictors of recurrent paralytic attacks[J].Eur J Neurol,2008,15(6):559-564.
[28]陈霞,郑荣秀,阚璇,等.儿童CACNA1S基因突变型低钾型周期性麻痹2例报告[J].临床儿科杂志,2015,33(12):1009-1012.
[29]Greig SL.Dichlorphenamide:a review in primary periodic paralyses[J].Drugs,2016,76(4):501-507.
R746.3
:A
:1004-583X(2017)03-0264-04
10.3969/j.issn.1004-583X.2017.03.021
2016-12-15 编辑:武峪峰
邹伟,Email:zouwei415@163.com
