对乙酰氨基酚与水分子间相互作用的密度泛函理论研究
2017-01-05黄茂胜黄嗣航张启航时军王芳
黄茂胜,黄嗣航,张启航,时军,王芳
(广东药科大学 中药学院,广东 广州 510006)
对乙酰氨基酚与水分子间相互作用的密度泛函理论研究
黄茂胜,黄嗣航,张启航,时军,王芳
(广东药科大学 中药学院,广东 广州 510006)
目的 探究对乙酰氨基酚与水分子之间的相互作用过程及其作用机制。方法 在DFT-B3LYP/6-311G(d p)水平下,求得对乙酰氨基酚(Ⅰ)与水(Ⅱ)二聚体(Ⅲ和Ⅳ)势能面上2种优化几何构型和电子结构,并进行能量及电荷分析。结果 经振动频率计算,确定2种二聚体(Ⅲ和Ⅳ)结构稳定,并求得它们在DFT-B3LYP/6-311G(d p)水平下的基组叠加误差(BSSE)分别为2.23、2.49 kJ/mol,ZPE校正能量分别为7.52、7.08 kJ/mol。经基组叠加误差和零点能校正后,对乙酰氨基酚与水二聚体分子间最大相互作用能为-51.95 kJ/mol。电荷分布与转移分析表明,两子体系间的电荷转移很多,接触点上氧原子和氢原子电荷变化较大。结论 对乙酰氨基酚与水分子形成二聚体时以氢键作用为主,且电荷和能量均有较大的变化,所形成的二聚体以Ⅲ较为稳定。
对乙酰氨基酚; 水; 二聚体; 密度泛函理论; 自然键轨道
对乙酰氨基酚(acetaminophen,以下简称AAP)分子式为C8H9NO2,相对分子质量151.16,熔点168~172 ℃,为临床常用的解热镇痛药[1]。其解热作用持久而缓慢,和阿司匹林相比,具有刺激性小、作用持久等特点。AAP常用的给药方式为口服给药,溶解性能是影响其口服生物利用度的重要物理化学参数。因此,了解AAP与水的作用机制,对提高AAP的水溶性具有重要意义。
2015年版《中国药典》中记载,AAP是一种白色、无臭,溶于甲醇、乙醇、丙酮和乙酸乙酯,易溶于热水,难溶于冷水的晶体,结构式见图1。由AAP的结构可以看出,AAP中的氨基(—NH2)和酚羟基(—OH)都可能与H2O分子形成分子间氢键。研究AAP-H2O超分子体系的结构和性能,比较分子之间的相互作用强弱,揭示相互作用的本质,对了解AAP与水的作用方式有重要意义[2-5]。
本文运用密度泛函理论(DFT)[6]和分子轨道杂化法(B3LYP)对AAP与H2O间相互作用的电子结构变化和相互作用能变化进行较系统地研究,旨在揭示AAP与H2O的作用规律,并期望对AAP的水溶解过程有所了解,进一步对如何提高AAP的水溶解度提供理论依据,同时对AAP-H2O新剂型的制备以及药动学的研究[7]提供指导。
1 计算方法
分子模拟的主要方法有经典力学模拟和量子力学模拟,而Gaussian一般适用于量子力学计算,其中量子力学模拟主要依据的方法有从头计算法、半经验分子轨道理论、密度泛函理论(DFT)方法等。
本文以Gview软件组建AAP单体C8H9NO2和H2O的二聚体C8H9NO2-H2O,在DFT-B3LYP/6-311G(d p)水平下[8],以本征值跟踪算法(EF)和内坐标的Berny算法等[9]对所选的结构进行全优化,求得能量极小点(稳定构型),经振动分析得优化结构无虚频。采用Boys和Bernardi提出的均衡校正法(CP)[10]进行基组叠加误差(BSSE)校正与零点能(ZPE)校正。通过自然轨道分析[11],揭示单体转变为二聚体后的电荷转移状况和分子间的相互作用,探索C8H9NO2和H2O形成二聚体时的相互作用的本质。全部计算均采用Guassian 09W程序[12]完成,收敛精度取程序内定值。
2 结果与讨论
2.1 单体的几何构型
AAP单体C8H9NO2和H2O的DFT-B3LYP/6-311G(d p)全优化几何构型及分子间距见图1,部分几何参数见表1。可见,经优化后的AAP单体呈平面构型,O11-H12的优化键长为0.096 3 nm,N13-H14的优化键长为0.100 8 nm。C1-O11-H12的键角大小为109.12°,C4-N13-H14和C15-N13-H14的键角大小分别为114.88°、116.26°。H23-O21-H22的优化键角为103.8°,这与H2O分子中2个H-O键的夹角为104.5°有差别,这可能是因为HF方法中没有充分考虑电子相关,以致所优化的水分子中的内氢键要弱一点。
表1 C8H9NO2和H2O的DFT-B3LYP/6-311G(d p)优化键长
Table 1 The optimized bond lengths of C8H9NO2and H2O at DFT-B3LYP/6-311G(d p)level 键长/nm


图1 AAP和H2O的全优化几何构型和部分原子编号
Figure 1 Optimized geometries and parts of atonmic numbering of APP and H2O
2.2 单体的电荷分布
自然集聚分析所得的AAP和H2O各原子上的净电荷见表2。可见,AAP中的氨基(—NH2)中的N13和H14所带电荷分别为-0.447e和0.223e,酚羟基(—OH)中的O11和H12所带电荷分别为-0.366e和0.247e,而水分子中O21、H22和H23所带电荷分别为-0.473e、0.237e、0.237e。
表2 C8H9NO2和H2O的DFT-B3LYP/6-311G(d p)自然原子电荷(e)
Table 2 The calculated natural atomic charges (e) of C8H9NO2and H2O at DFT-B3LYP/6-311G (d p) level

原子AAP(Ⅰ)H2O(Ⅱ)C10.155C2-0.092C3-0.105C40.162C5-0.085C6-0.133H70.103H80.087H90.140H100.091O11-0.366H120.247N13-0.447H140.223C150.320O16-0.370C17-0.307H180.141H190.095H200.095O21-0.473H220.237H230.237
2.3 二聚体的几何结构
二聚体C8H9NO2-H2O(Ⅲ和Ⅳ)的DFT-B3LYP/6-311G(d p)全优化几何构型及分子间距见图2。可见,在对乙酰氨基酚与水的2种二聚体中均只有1个接触点,而且二聚体Ⅲ中的接触点(O-H…O)距离为0.182 3 nm,短于Ⅳ中的接触点(N-H…O)距离0.198 9 nm。由表3中的部分几何参数可见,图2中的Ⅲ和Ⅳ均近似地属于C1点群,与单体相比,接触点为O-H…O的二聚体Ⅲ中的O-H键长变化较大(Ⅲ增长0.001 1 nm),接触点为N-H…O的二聚体Ⅳ中的N-H键长变化较小(Ⅳ增长0.007 0 nm)。与单体相比,2种二聚体键角变化均小于0.5°,二面角变化均小于0.2°。由此可见,聚合和分子间相互作用未造成各单体较大的扭曲,对内旋转影响也比较小。根据二聚体的接触点种类和距离可以初步推测出Ⅲ的稳定性优于Ⅳ。
表3 二聚体C8H9NO2-H2O的DFT-B3LYP/6-311G(d p)优化键长
Table 3 The optimized bond lengths of C8H9NO2-H2O at DFT-B3LYP/6-311G (d p) level

键长/nm
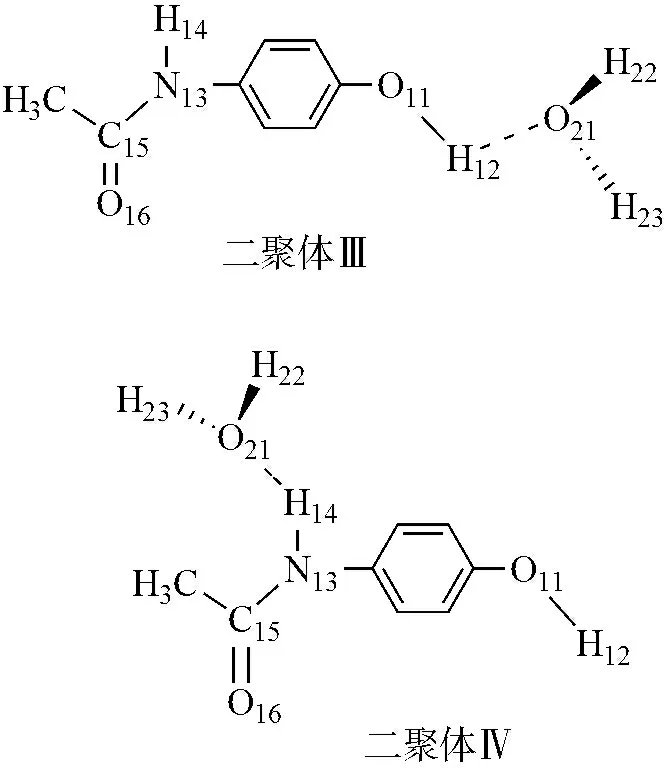
图2 二聚体C8H9NO2-H2O的全优化几何构型和部分原子编号
Figure 2 Optimized geometries and parts of atonmic numbering of C8H9NO2-H2O
2.4 二聚体的电荷分布和转移
自然集聚分析所得的AAP和水的2种二聚体各原子上的净电荷见表4。结合表4、图1和图2可见,与单体相比,二聚体上的各原子电荷变化不大,但是在接触点上的氧原子、氮原子和氢原子及附近的氧原子变化明显。如Ⅲ中的O11为-0.394e减少0.028e,Ⅳ中的N13为-0.466e减少0.019e,而H14为0.263e相比单体增加了0.040e,结合后H2O上的氧原子和氢原子的电荷也有明显变化。子体系间电荷转移比较多,两子体系间电荷传递主要通过接触点O-H…O和N-H…O传递,电荷分别为0.078e和0.081e。
表4 二聚体C8H9NO2-H2O的DFT-B3LYP/6-311G(d p)自然原子电荷(e)
Table 4 The calculated natural atomic charges(e)of C8H9NO2-H2O at DFT-B3LYP/6-311G (d p) level
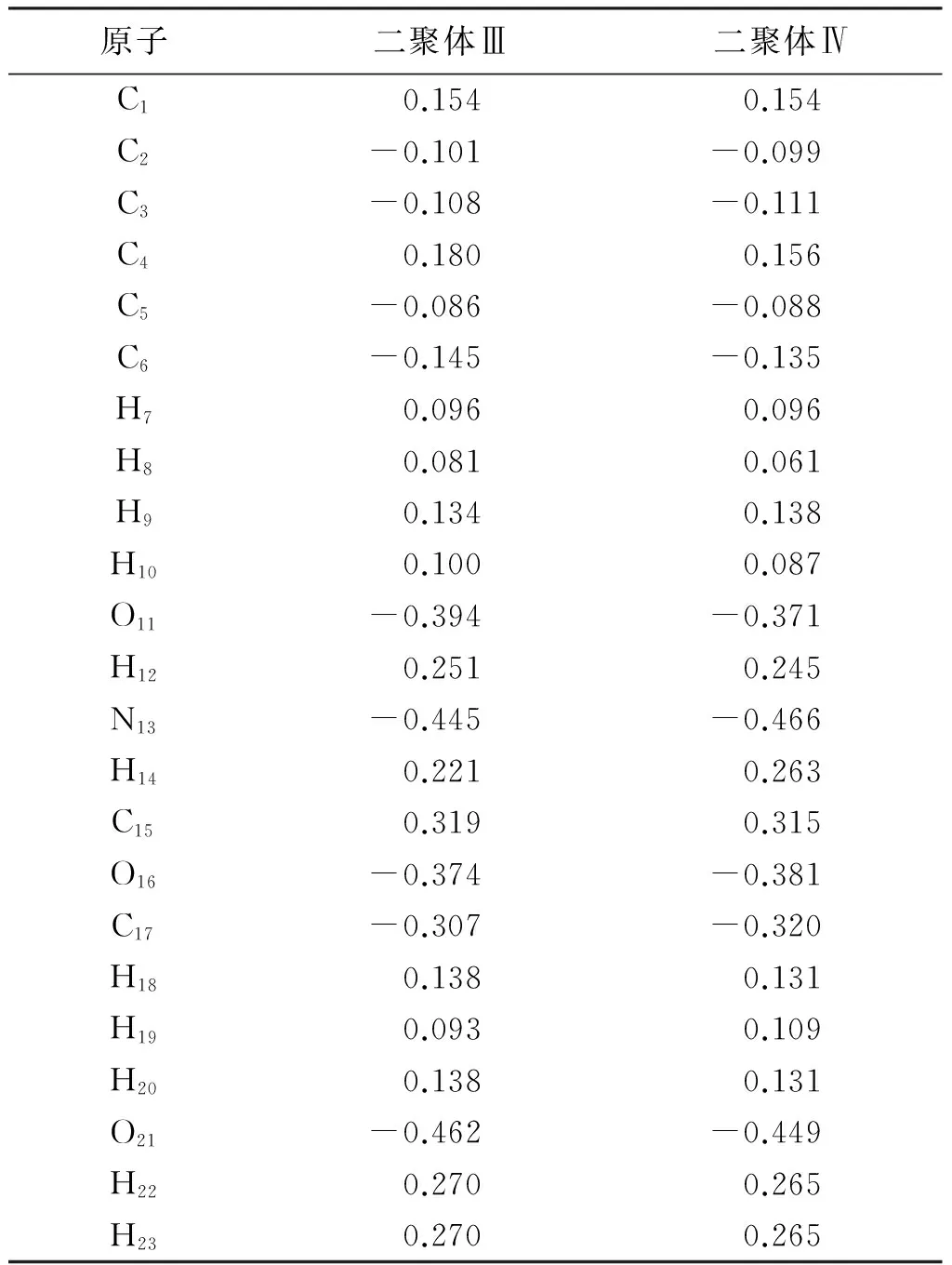
原子二聚体Ⅲ二聚体ⅣC10.1540.154C2-0.101-0.099C3-0.108-0.111C40.1800.156C5-0.086-0.088C6-0.145-0.135H70.0960.096H80.0810.061H90.1340.138H100.1000.087O11-0.394-0.371H120.2510.245N13-0.445-0.466H140.2210.263C150.3190.315O16-0.374-0.381C17-0.307-0.320H180.1380.131H190.0930.109H200.1380.131O21-0.462-0.449H220.2700.265H230.2700.265
2.5 相互作用能
在DFT-B3LYP/6-311G(d p)水平下,求得的AAP和水形成二聚体时的分子间相互作用能见表5,包括DET总能量(EDET)、零点能(ZPE)、相互作用能(ΔEDET)、经基组叠加误差(BSSE)和ZPE校正后的(ΔE)C4部分。对比未校正的相互作用能ΔEDET,大小顺序为:Ⅲ﹥Ⅳ。2种二聚体在DFT-B3LYP/6-311G(d p)水平下的BSSE分别为2.23 kJ/mol和2.49 kJ/mol,ZPE校正值分别为7.52 kJ/mol和7.08 kJ/mol。这表明进行BSSE和ZPE校正都是必要的。对比经BSSE和ZPE校正后的相互作用能(ΔE)C,大小顺序为:Ⅲ﹥Ⅳ,结果与ΔEDET一致。经BSSE和ZPE校正后的AAP与水二聚体的最大相互作用能为-51.95 kJ/mol,属于构型Ⅲ(构型Ⅳ的相互作用能为-47.92 kJ/mol),这与前面根据键长和接触点距离来推测二聚体稳定性基本一致。
表5 DFT-B3LYP/6-311G(d p)的总能量、零点能和相互作用能
Table 5 Total energies,zero point energy and binding energies at DFT-B3LYP/6-311G (d p) levelE/(kJ·mol-1)

2.6 自然键轨道分析
在DFT-B3LYP/6-311G(d p)水平下,对AAP和水形成的二聚体进行自然键轨道(NBO)分析,电子供体轨道与电子受体轨道之间的相互作用稳定化能ΔE见表6。可见,在构型Ⅲ中,O21的孤对电子(2)对O11-H12的e反键轨道的稳定化能为99.60 kJ/mol。从而表明,在二聚体Ⅲ的子体系之间,相互作用主要发生在Ⅲ中的O21的孤对电子(2)与O11-H12的e反键轨道之间。而在Ⅳ中,N13-H14共价键对O21-H23的e反键轨道稳定化能为73.74 kJ/mol,表明在二聚体Ⅳ的子体系之间,相互作用主要发生在Ⅳ中的N13-H14共价键与O21-H23的e反键轨道之间。
表6 二聚体C8H9NO2-H2O的DFT-B3LYP/6-311G(d p)自然键轨道分析部分结果
Table 6 The calculated results of C8H9NO2-H2O at the DFT-B3LYP/6-311G(d p)level by NBO analysis

二聚物供电子体NBO电子受体NBOΔE/(kJ·mol-1)ⅢLP(1)O21BD*(1)O11-H120.59ⅢLP(2)O21BD*(1)O11-H1299.60ⅣBD(1)N13-H14BD*(1)O21-H220.46ⅣBD(1)N13-H14BD*(1)O21-H2273.74ⅣLP(1)O21BD*(1)N13-H140.54
注: ΔE为稳定化能,BD为成键轨道,BD*为反键轨道,LP为非公有电子对;对于BD和BD*中,(1)与(2)各自代表σ轨道和π轨道;对于LP中,(1)和(2)分别表示第1孤对电子和第2孤对电子。
稳定化能ΔE越大,表示供电子体(donor)与电子受体(acceptor)的相互作用越强。Ⅲ和Ⅳ最大的稳定化能分别为99.60 kJ/mol和73.74 kJ/mol,大小顺序为:Ⅲ﹥Ⅳ,这与前面的结合能变化规律及根据键长和接触点距离推测二聚体稳定性的结果基本一致。
3 结论
在DFT-B3LYP/6-311G(d p)水平下,求得AAP与水的2种二聚体均有1个分子间氢键,且经振动分析得优化结构无虚频,具有稳定的结构。Ⅲ中的接触点(O-H…O)距离为0.182 3 nm,短于Ⅳ中的接触点(N-H…O)距离(0.198 9 nm)。2种AAP的二子体系之间电荷转移很多,接触点上的氧原子、氮原子和氢原子及附近的氧原子电荷变化明显。在二聚体Ⅲ的子体系之间,相互作用主要发生在O21的孤对电子与O11-H12的e反键轨道之间,而在二聚体Ⅳ的子体系之间,相互作用主要发生在N13-H14共价键与O21-H23的e反键轨道之间。最大的相互作用能为-51.95 kJ/mol,属于构型Ⅲ,而构型Ⅳ的相互作用能为-47.92 kJ/mol。
[1] 王柳萍,杨斌.对乙酰氨基酚药动学研究进展[J].时珍国医国药,2011,22(2):469-470.
[2] 陈鹏宇,张旭,吴峰,等.TiO2光催化降解水中对乙酰氨基酚的研究[J].安全与环境工程,2007,14(3):40-42.
[3] 张学慧,王国年.对乙酰氨基酚在术后镇痛中的应用[J].临床麻醉学杂志,2016(2):198-200.
[4] 黄辉,董海山.一类对撞击不敏感的新型炸药[J].含能材料,2002,10(2):74-77.
[5] JOLLOW D J,THORGEIRSSON S S,POTTER W Z,et al. Acetaminophen[J]. Pharmacology,1974,12(4/5):251-271.
[6] KOHN W,SHAM L J. Self-consitent equations including exchange and correlation effects[J]. Phys Rev A,1965,140:1133-1138.
[7] 王静,王华丽,臧恒昌.对乙酰氨基酚合成方法的研究进展[J].食品与药品,2010,12(5):354-356.
[8] 张俊吉.对乙酰氨基酚和壳聚糖的结构和光谱研究[D].郑州:河南大学,2008.
[9] MENCH M M,KUO K K,YEH C L,et al. Comparison of thermal behavior of regular and ultra-fine aluminum powders (Alex) made from plasma explosion process[J]. Combust Sci Technol,1998,135(1):269-292.
[10] 黄辉,黄勇,李尚斌.含纳米级铝粉的复合炸药研究[J].火炸药学报,2002,25(2):1-3.
[11] FATHAUER R W,GEORGE T,KSENDZOV A,et al. Visible luminescence from silicon wafers subjected to stain etches[J]. Appl Phys Lett,1992,60(8):995-997.
[12] YU Weifei,HU Hui,NIE F D,et al. Research on nano-composite energetic materials[J]. Energet Mater,2005,38(2):256-268.
(责任编辑:陈翔)
Study on the density function theory on intermolecular interaction of acetaminophen and water
HUANG Maosheng,HUANG Sihang,ZHANG Qihang,SHI Jun,WANG Fang
(SchoolofTraditionalChineseMedicine,GuangdongPharmaceuticalUniversity,Guangzhou510006,China)
Objective To study the interaction process between acetaminophen and water molecules as well as its underlying mechanism. Methods The optimized geometries and electronic structures of acetaminophen (Ⅰ),water (Ⅱ) and their possible stable dimers (Ⅲ and Ⅳ) were obtained at the DFT-B3LYP/6-311G (dp) level. Energy and charge were also measured. Results The stabilization of the dimers was ascertained by vibrational frequency calculation. The values of the Basis Set Superposition Error (BSSE) at DFT-B3LYP/6-311G (d p) level were 2.23 kJ/mol and 2.49 kJ/mol for Ⅲ and Ⅳ,respectively. The zero point energy (ZPE) corrections for the interaction energies were 7.52 kJ/mol and 7.08 kJ/mol for Ⅲ and Ⅳ,respectively. After BSSE and ZPE corrections,the maximum corrected binding energy of acetaminophen-water dimers reached -51.95 kJ/mol. In addition,charge distribution and transfer analysis showed that charge redistribution mainly occurred on the adjacent O-H…O atoms of acetaminopen dimers between submolecules while the change of the charge between two submolecules was small for all two acetaminophen-water dimers. Conclusion Hydrogen bonding is the main effect on the formation of acetaminophen-water dimers whose energy and charge are great changes and the constitution of Ⅲ is the most stable.
acetaminophen;water; dimmer; density functional theory; natural bond orbital
2016-09-12
广东省科技计划项目(2013B021800088);广东省医学科研基金项目(B2014209)
黄茂胜(1993—),男,2016级硕士研究生,Email:13631194354@163.com;通信作者:黄嗣航(1979—),副教授,硕士研究生导师,从事药物新剂型与新技术研究,电话:020-39352169,Email:sihanghuang@yeah.net。
时间:2016-11-29 10:57
http://www.cnki.net/kcms/detail/44.1413.R.20161129.1057.006.html
R913
A
1006-8783(2016)06-0712-05
10.16809/j.cnki.1006-8783.2016091202
