三疣梭子蟹PtCrustin2成熟肽在毕赤酵母中的表达
2017-01-03王艳慧
王艳慧, 陶 妍
(上海海洋大学食品学院 上海水产品加工及贮藏工程技术研究中心,上海 201306)
三疣梭子蟹PtCrustin2成熟肽在毕赤酵母中的表达
王艳慧, 陶 妍*
(上海海洋大学食品学院 上海水产品加工及贮藏工程技术研究中心,上海 201306)
Crustin主要分布于甲壳动物中,是一种富含半胱氨酸的小分子抗菌肽,在甲壳动物的先天免疫系统中发挥重要作用。根据crustin的一级结构特征可以将其分为不同的类型,本文以三疣梭子蟹(Portunustrituberculatus)的PtCrustin2成熟肽为研究对象,通过构建毕赤酵母表达系统,以期实现PtCrustin2成熟肽在毕赤酵母中的重组DNA表达。首先,从其鳃中提取总RNA,通过RT-PCR得到编码PtCrustin2成熟肽的cDNA(mPtCrustin2),并在其5′和3′端分别引入EcoRⅠ和NotⅠ限制性内切酶位点;然后将此片段与表达载体pPIC9K连接,构建重组表达载体pPIC9K-mPtCrustin2;电转入毕赤酵母GS115细胞后,以不同浓度的G418筛选到高拷贝酵母转化子,经0.5%甲醇诱导表达和固化金属离子亲和层析(IMAC)分离,获得了纯化的重组体mPtCrustin2,Tricine-SDS-PAGE分析显示其分子量约10.5 kDa;抑菌实验证明重组体mPtCrustin2对金黄色葡萄球菌和铜绿假单胞菌具有一定的抑菌效果。本研究首次实现了三疣梭子蟹PtCrustin2成熟肽在毕赤酵母中的重组DNA表达,为进一步研究提供参考。
三疣梭子蟹;mPtCrustin2成熟肽;毕赤酵母;重组DNA表达
Crustin主要分布于甲壳动物中,是一类富含半胱氨酸的阳离子抗菌肽,分子量为7~14 kDa;N端含有一个信号序列,C端含有一个WAP(whey acidic protein)结构域,该结构域包含8个保守的半胱氨酸残基,形成一个含有4个二硫键的紧密包裹结构,是crustin的主要功能域[1]。信号序列与WAP结构域之间为可变区域,Smith等[2]根据可变区域的结构差异,将crustin分为三种类型:Ⅰ型、Ⅱ型和Ⅲ型。Ⅰ型crustin中,可变区域富含半胱氨酸;Ⅱ型crustin可变区域不仅富含半胱氨酸,且在信号序列附近有约40~80个甘氨酸;Ⅲ型crustin在信号序列与WAP结构域之间含有一个短的富含PRP(脯氨酸-精氨酸-脯氨酸)的区域。Crustin作为一种抗菌因子在甲壳动物先天性免疫应答中发挥了重要作用,第一个crustin是在普通滨蟹(Carcinusmaenas)血细胞中分离到的,命名为carcinin,属于Ⅰ型crustin,对革兰阳性菌有抑菌活性[3]。近年来研究者对来自三疣梭子蟹(Portunustrituberculatus)血细胞和眼柄的I型crustin的3个同工型(PtCrustin1、PtCrustin2、PtCrustin3)进行体外抑菌试验,发现它们不仅对革兰阳性的藤黄微球菌(Micrococcusluteus)和金黄色葡萄球菌(Staphylococcusaureus)有抑菌活性,而且对革兰阴性的铜绿假单胞菌(Pseudomonasaeruginosa)也有抑菌活性[4]。PtCrustin2主要在三疣梭子蟹的鳃和眼柄中表达,Cui等[4]克隆了三疣梭子蟹PtCrustin2的成熟肽基因,并通过大肠埃希菌原核表达系统获得了重组体PtCrustin2成熟肽。然而,迄今为止,关于crustin重组DNA表达方面的研究报道很少,尤其是基于真核重组表达的PtCrustin2的制备未见报道。本研究以三疣梭子蟹包含WAP结构域的PtCrustin2成熟肽为研究对象,首先通过RT-PCR从三疣梭子蟹的鳃中克隆到编码mPtCrustin2的cDNA片段,经巢式PCR对其添加EcoRⅠ和NotⅠ限制性内切酶位点和6×his标签;以pPIC9K作为表达载体,构建真核重组表达载体pPIC9K-mPtCrustin2;电转至毕赤酵母GS115后,通过甲醇诱导表达重组体mPtCrustin2,采用固化金属离子亲和层析(IMAC)法对其进行纯化和鉴定,并通过抑菌实验对其生物学活性进行验证。
1 材料与方法
1.1 材料
1.1.1 质粒和菌株 pMD19-T质粒购自Takara公司(日本),大肠埃希菌DH5α为本实验室保存菌株,pPIC9K表达载体和毕赤酵母GS115购自Invitrogen公司(美国)。
1.1.2 主要试剂 RNAiso plus、TaqDNA聚合酶、EcoRⅠ和NotⅠ均购自Takara公司;DNA回收试剂盒、质粒提取试剂盒、酵母基因组提取试剂盒、DNA分子量Marker和蛋白质分子量标准均购自天根生化科技有限公司(北京),引物合成和DNA测序由上海生物工程有限公司完成。
1.2 方法
1.2.1 提取总RNA和合成第一股cDNA 以三疣梭子蟹的鳃为材料,按照RNAiso Plus说明书提取总RNA。第一股cDNA合成如下操作:6 μL总RNA、1 μL Oligo dT Primer、1 μL dNTP、2 μL RNase free ddH2O,65 ℃保温5 min,冰上迅速冷却;依次加入4 μL 5×Prime Script Buffer、0.5 μL RNase Inhibiter、1 μL Prime Script RTase、4.5 μL RNase free ddH2O缓慢混匀;42 ℃保温60 min;95 ℃保温5 min。
1.2.2 PtCrustin2成熟肽基因的PCR扩增和克隆 参考三疣梭子蟹PtCrustin2的全长cDNA序列(GenBank: JQ728435),设计三对引物(表1),通过巢式PCR(图1),得到添加EcoRⅠ、NotⅠ酶切位点和6×his标签的PtCrustin2成熟肽基因。3次PCR的反应条件如下:10×PCR Buffer 2 μL、dNTP 1.6 μL、正向和反向引物各0.8 μL、DNA模板0.5 μL、TaqDNA聚合酶(2.5 U/μL)0.2 μL,无菌水加至20 μL;94 ℃变性3 min; 94 ℃预变性30 s,56 ℃(60 ℃)(61 ℃)退火30 s,72 ℃延伸1 min,共30个循环; 72 ℃延伸5 min。第3次PCR产物割胶纯化后与克隆载体pMD-19T进行连接,得到重组质粒pMD-19T-mPtCrustin2,将其转至大肠埃希菌DH5α感受态细胞中,37 ℃培养过夜,通过菌落PCR挑取阳性克隆,送至上海生工生物工程有限公司进行DNA测序。
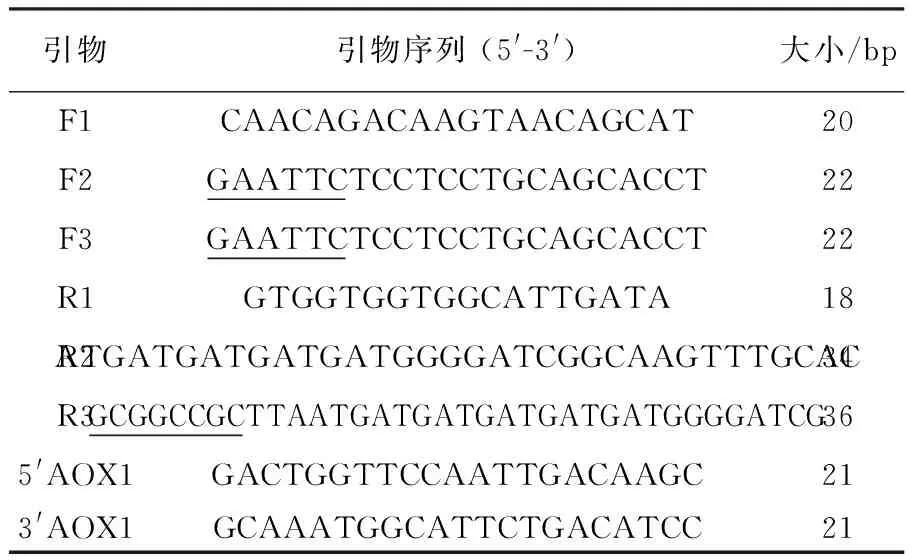
表1 引物序列Table 1 Sequences of primers
注:下划线表示EcoRⅠ和NotⅠ的酶切位点
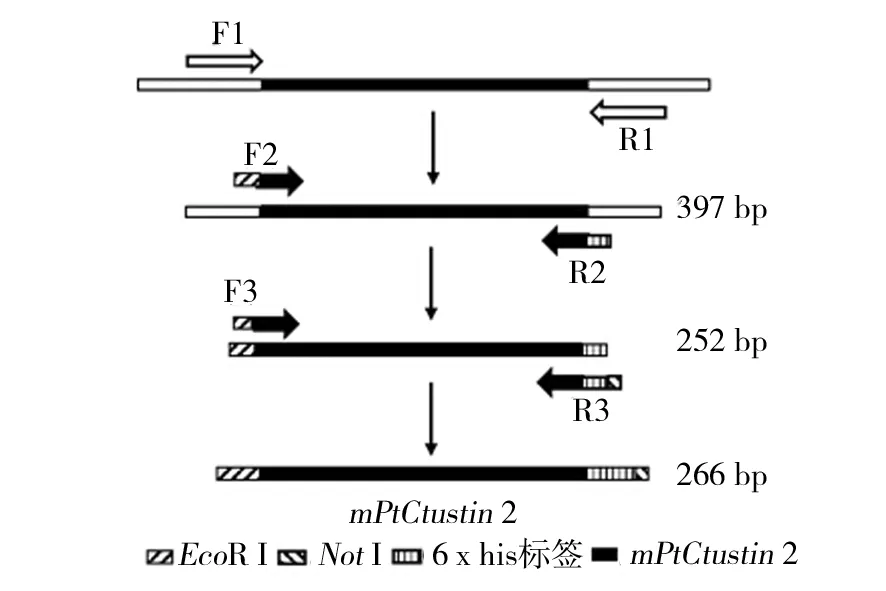
图1 通过PCR扩增目的基因的策略图Fig.1 Strategic map for PCR amplification of target gene
1.2.3 pPIC9K-mPtCrustin2重组表达载体的构建及鉴定 使用EcoRⅠ和NotⅠ对pMD-19T-mPtCrustin2进行双酶切,得到含酶切位点的目的片段,将其与用同样酶处理过的表达载体pPIC9K按1∶4(体积比)混合,在T4 DNA连接酶作用下,16 ℃反应16 h后,转化至大肠埃希菌DH5α感受态细胞中,37 ℃培养过夜。通过菌落PCR、EcoRⅠ和NotⅠ双酶切、DNA测序鉴定重组表达载体pPIC9K-mPtCrustin2是否构建成功。
1.2.4 电转入毕赤酵母及高拷贝酵母转化子的筛选 用SacⅠ对重组表达载体pPIC9K-mPtCrustin2进行线性化处理,产物经割胶纯化后以1∶8(体积比)与毕赤酵母GS115感受态细胞混合,转入预冷的0.2 cm电转杯中,冰浴5 min;1.5 kV、25 μF、200 Ω,电击5 ms,立即加入1 mL预冷的1 mol/L山梨醇;30 ℃静置2 h后,离心将菌体涂布于MD平板,30 ℃孵育至单菌落产生。挑取88个单菌落分别接种在含有0~4.0 mg/mL G418的YPD平板上,筛选高拷贝转化子;将筛选到的转化子转接到MM平板和MD平板上,筛选甲醇利用快速型转化子,然后接种于YPD液体培养基中培养。提取酵母基因组DNA,并以此为模板,采用pPIC9K的通用引物5′AOX1和3′AOX1(表1)进行PCR鉴定。
1.2.5 重组体mPtCrustin2的诱导表达及分离纯化和Western blot鉴定 将上述筛选到的高拷贝转化子在BMG培养基中使用0.5%甲醇,30 ℃、250 r/min培养至OD600为2~6;离心收集菌体,重悬于BMM培养基中,28 ℃、250 r/min培养96 h,期间每隔24 h补加甲醇至终体积的0.5%;离心收集上清,用作Tricine-SDS-PAGE分析(浓缩胶浓度4%、夹层胶浓度10%、分离胶浓度15.5%)和纯化,蛋白质分子量标准由中科瑞泰生物科技有限公司(北京)提供。另一方面,将上清过0.22 μm滤膜后,采用赛多利斯切向流超滤器(VIVAFLOW 200)对其进行浓缩脱盐,再采用固化金属离子亲和层析(IMAC)进行纯化,纯化产物溶于PBS缓冲液(pH=7.0)。纯化的目的蛋白经Tricine-SDS-PAGE后,将凝胶电转至PVDF膜(100 V,1.5 h),用TBST漂洗后加入封闭液封闭1 h;再用TBST漂洗后加入抗his标签鼠单克隆抗体,室温孵育2 h;再用TBST漂洗后加入辣根过氧化物酶标记山羊抗小鼠IgG (H+L),室温孵育1 h;最终经TBST漂洗后用DAB显色液试剂盒进行显色。
1.2.6 重组体mPtCrustin2的活性检测 以金黄色葡萄球菌和铜绿假单胞菌为供试菌株,在LB液体培养基中,37 ℃、150 r/min培养至对数生长期;取1 μL上述菌液,稀释至50 μL,分别与50 μL纯化目的蛋白和50 μL PBS缓冲液(阴性对照)混匀,37 ℃培养12 h,将此培养液涂于LB平板上,37 ℃培养12 h后,观察菌落情况。
2 结果与分析
2.1 RT-PCR获得目的基因及其克隆测序
提取三疣梭子蟹鳃的总RNA后,通过反转录合成第一股cDNA,并以此为模板进行巢式PCR。如图2所示,第1次PCR得到含mPtCrustin2序列及部分非编码序列的397 bp的片段(图2A);第2次PCR得到5′端和3′端分别含EcoRⅠ和5×his标签的252 bp 的mPtCrustin2片段(图2B);第3次PCR得到5′端和3′端分别含EcoRⅠ、NotⅠ酶切位点、终止密码子(TAA)和6×his标签的266 bp的目的片段(图2C)。DNA测序结果与基因库序列一致(图3A),由推断的氨基酸序列可知,PtCrustin2成熟肽包含12个半胱氨酸残基,在空间上可以形成6对二硫键,以维持其结构的稳定和生物学功能。
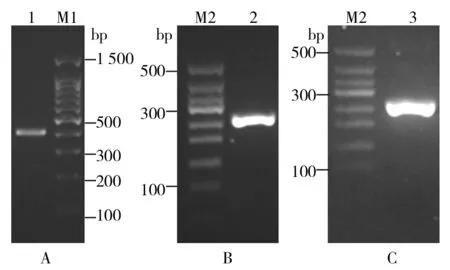
图2 目的基因的PCR扩增Fig.2 PCR amplification of target gene M1、M2:DNA 分子量标准;1、2、3:3次PCR扩增的产物 M1,M2:DNA marker; 1, 2 and 3:PCR products for the first, second and third time respectively
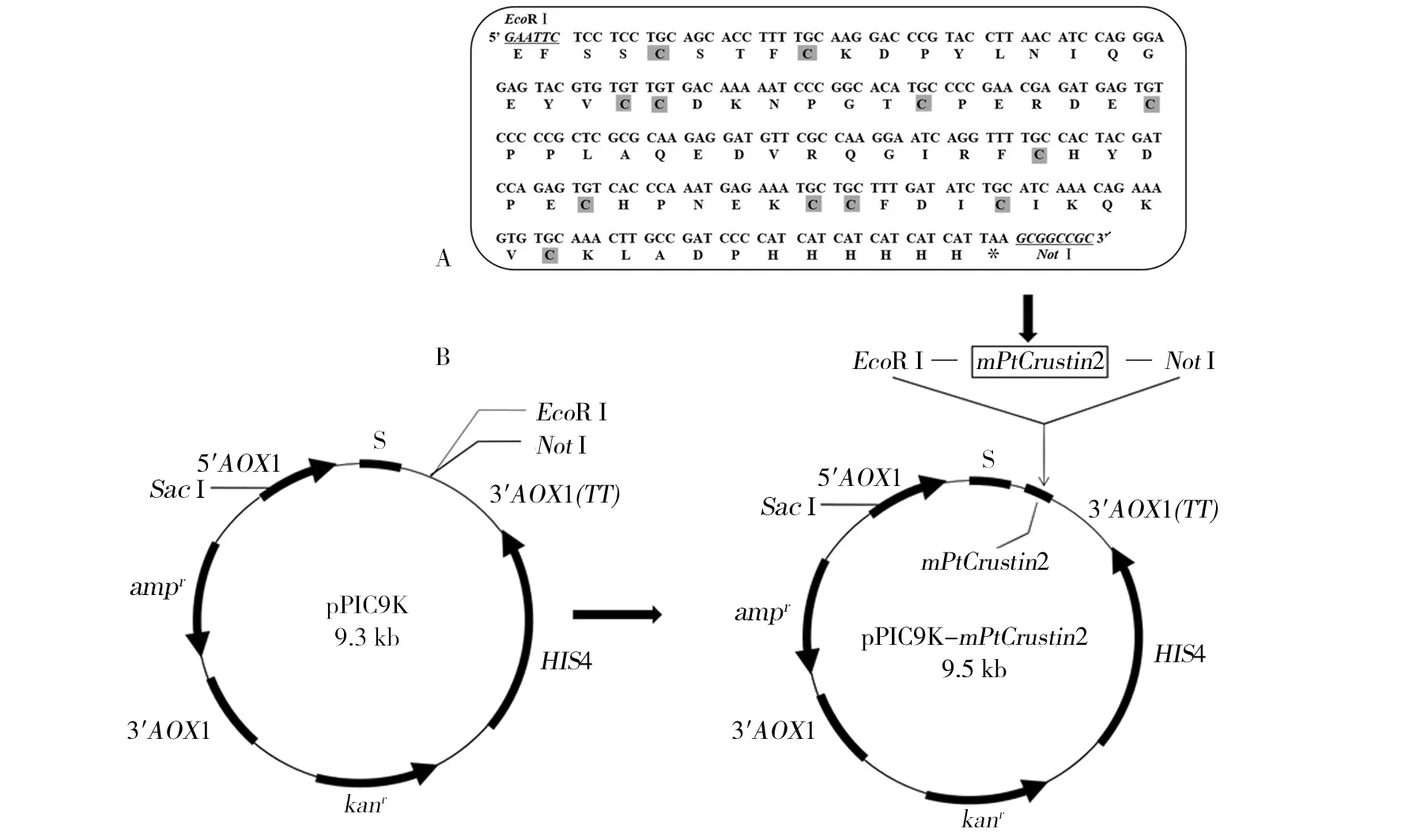
图3 mPtCrustin2的cDNA和推断的氨基酸序列(A)及其重组表达载体pPIC9K-mPtCrustin2的构建(B)Fig.3 cDNA and deduced amino acid sequences of mPtCrustin2 (A) and construction of the recombinant expression vector pPIC9K-mPtCrustin2 (B) *为终止密码子,下划线为限制性酶切位点,阴影为保守的半胱氨酸残基 Stop code is shown by star, restriction endonucleases are underlined, and cysteine residues are shaded
2.3 构建重组表达载体pPIC9K-mPtCrustin2及其鉴定
采用EcoRⅠ和NotⅠ对上述重组质粒pMD-19T-mPtCrustin2和表达载体pPIC9K同时进行双酶切后,将mPtCrustin2片段与线性的pPIC9K连接,构建pPIC9K-mPtCrustin2重组表达载体(图3B);转化至大肠埃希菌DH5α感受态细胞后,使用来自于目的片段上的引物(F3)和载体上的3′AOX1引物进行菌落PCR鉴定,以考察两者是否连接成功。由图4A可见,在约372 bp处有明显条带,与理论值相符;另一方面,从大肠埃希菌中提取重组质粒pPIC9K-mPtCrustin2并进行双酶切验证,由图4B可见大、小分子量的2个条带,位于较低位置处的条带是mPtCrustin2,大小约为266 bp,另一个大分子量的条带来自于载体。此外,DNA测序结果也证明mPtCrustin2与pPIC9K正确连接,未发生基因变异。
2.4 高拷贝酵母转化子的筛选及其鉴定
将重组表达载体pPIC9K-mPtCrustin2电转入毕赤酵母GS115细胞后,经含不同浓度G418的YPD平板、MM平板和MD平板筛选后,获得8株甲醇利用快速型高拷贝转化子;经YPD液体培养后,提取酵母基因组DNA,并以此为模板,使用载体上的通用引物5′AOX1和3′AOX1,通过PCR考察重组表达载体pPIC9K-mPtCrustin2是否与酵母基因组DNA嵌合。如图5所示, 1~8是以含pPIC9K-mPtCrustin2的酵母基因组DNA为模板的PCR产物,9是以含pPIC9K空载体的酵母基因组DNA为模板的PCR产物。1~8号样品均在约739 bp处有明显条带,与理论值相符;而9号样品作为阴性对照,在约493 bp处有明显条带,由此证明pPIC9K-mPtCrustin2成功嵌合进毕赤酵母基因组DNA。
2.5 在毕赤酵母中表达目的蛋白及其表达产物的纯化和Western blot鉴定
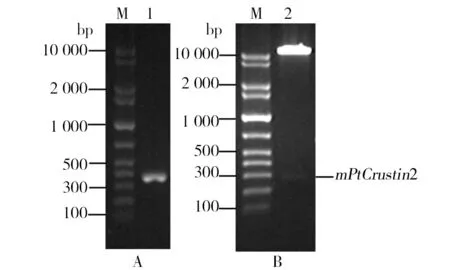
图4 重组表达载体pPIC9K-mPtCrustin2的菌落 PCR (A)和双酶切鉴定(B)Fig.4 Colony PCR (A) and restriction endonuclease digestion (B) identifications for the recombinant expression vector pPIC9K-mPtCrustin2M:DNA 分子量标准;1:菌落PCR产物;2:EcoRⅠ和NotⅠ双酶切产物M:DNA marker; 1:colony PCR product; 2:products digested by EcoRⅠand NotⅠ
将筛选到的高拷贝酵母转化子在BMM培养基中培养,期间不断补加甲醇使其终浓度为0.5%,诱导表达96 h后,通过离心分离收集上清。由Tricine-SDS-PAGE分析可见,在14.4~7.8kDa处有2个条带(图6A),位于下方的那个条带为mPtCrustin2,分子质量约为10.5 kDa,符合表达产物的理论分子质量,该表达产物因含限制性酶切位点和6×his标签以及信号肽酶切位点,故较纯粹的mPtCrustin2的分子质量略高;而含pPIC9K空载体的酵母转化子的培养液上清未发现清晰条带。进一步将收集的上清通过切向流超滤仪对其进行浓缩脱盐后采用IMAC层析法分离纯化目的蛋白,如图6B所示, 在14.4~7.8 kDa处有单一明显条带,与图6A中显示的目的蛋白位置相同,证明已获得较高纯度的目的蛋白。纯化的15 mL蛋白质溶液的浓度为0.35 mg/mL,推算其表达量为5.25 mg/L。另一方面,Western blot分析显示(图7),在约10.5 kDa处有明显条带,与预测理论值相符,进一步证明了目的蛋白被成功表达。
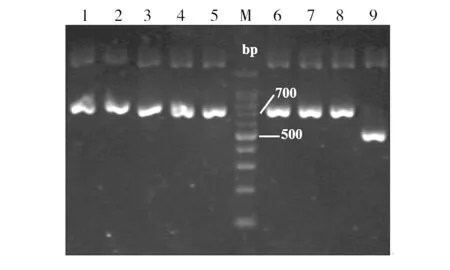
图5 高拷贝酵母转化子的PCR鉴定Fig.5 PCR identification for transformants containing multicopy gene insertionM:DNA 分子量标准;1~8:含pPIC9K-mPtCrustin2的酵母转化子;9:含pPIC9K的酵母转化子M:DNA marker;1~8:yeast transformants containing pPIC9K-mPtCrustin2; 9:yeast transformants containing pPIC9K
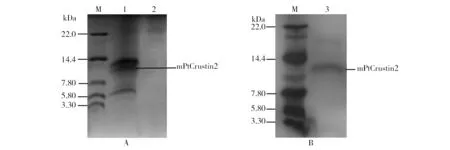
图6 培养液上清(A)和纯化产物(B)的Tricine-SDS-PAGE分析Fig.6 Tricine-SDS-PAGE analysis for the supernatant (A) and purified productM:蛋白质分子量标准;1:培养96 h的上清;2:阴性对照;3:纯化后的产物M:protein marker; 1:supernatant cultured for 96 h; 2:negative control; 3:purified product

图7 纯化产物的Western blot分析Fig.7 Western blot analysis for the purified productM:蛋白质分子量标准;1:纯化后的产物;2:阴性对照M:protein marker; 1:purified product; 2:negative control
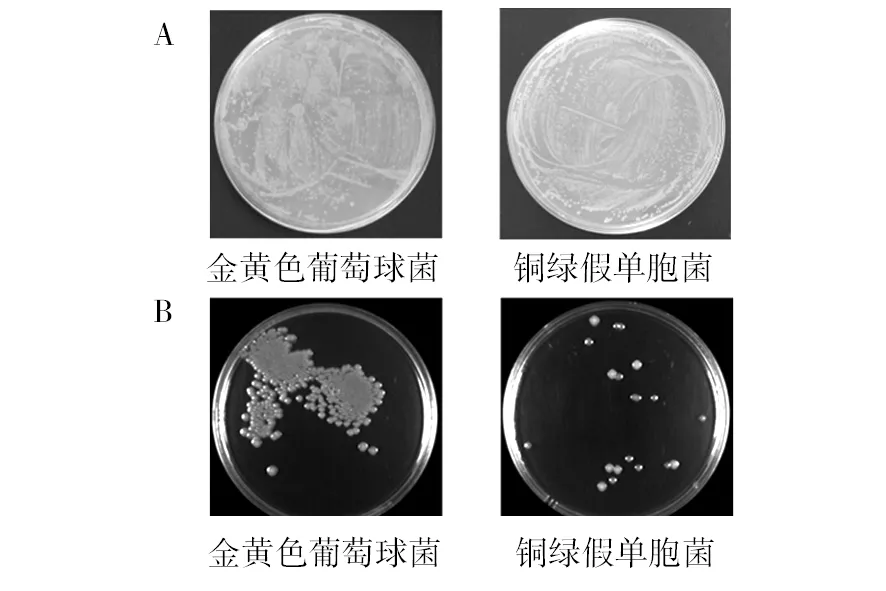
图8 重组体mPtCrustin2的抑菌活性Fig.8 Antimicrobial activity of recombinant mPtCrustin2A:阴性对照;B:重组体mPtCrustin2A:negative control; B:recombinant mPtCrustin2
2.6 重组体mPtCrustin2的抑菌活性
抑菌实验结果如图8所示,A为PBS缓冲液(阴性对照),2个平板上长满菌落;而B图的2个平板显示重组体mPtCrustin2对革兰阳性的金黄色葡萄球菌和革兰阴性的铜绿假单胞菌均有抑菌活性,但对后者的抑菌效果更好。
3 讨 论
迄今为止,关于三疣梭子蟹PtCrustin2的重组DNA表达研究仅有原核表达的报道,且表达量很低[4]。本研究选择毕赤酵母表达系统是基于其表达稳定性高,能高效表达外源蛋白并具有翻译后修饰功能,故有利于获得具生物学活性的重组蛋白[5],旨在为进一步的扩大制备和功能研究奠定基础。通过构建pPIC9K-mPtCrustin2真核重组表达载体,初步实现了三疣梭子蟹mPtCrustin2在毕赤酵母中的重组DNA表达,虽然表达量不高,但经抑菌实验初步确证重组体mPtCrustin2具有一定的抑菌活性。关于重组表达载体的构建方面还有待进一步的改进,载体pPIC9K中α因子信号肽的N端存在Kex2和Ste13两个切割位点,由于目的片段5′端所添加的限制性内切酶位点为EcoRⅠ,而EcoRⅠ与Kex2之间的序列导致重组体mPtCrustin2 N末端额外引入6个氨基酸;另外,Ste13因被切割效率不高,可能导致重组体的N末端氨基酸残基稍有差异[6],因此并未获得天然的N末端。至于N末端额外引入的氨基酸是否对目的蛋白的空间结构以及生物学活性产生影响还有待于进一步的研究。
已有研究表明,对密码子进行优化后可以提高目的蛋白的表达量[7-9]。由于本研究使用的编码mPtCrustin2的基因是通过RT-PCR获得的天然cDNA,对于毕赤酵母来说可能存在某些密码子的限制,这可能是导致目的蛋白表达量不太高的主要原因。经检测,96个密码子中低频密码子的比例为6%,如天冬氨酸、谷氨酸、甘氨酸等都是由低频密码子编码,因此进一步的研究将根据毕赤酵母的密码子偏爱性,对目的基因进行密码子优化,以期提高目的蛋白的表达量。另外,拟选择pPICZαA替代pPIC9K,因pPICZαA分子量较小,长度仅为pPIC9K的1/3,转化操作和染色体的整合相对容易[10],通过采用合适的限制性内切酶,可以获得具有天然N末端的重组体mPtCrustin2。另一方面,以野生型的、对外源蛋白具有较好分泌和适应能力的毕赤酵母X-33作为工程菌[11],以期获得高表达量的、具良好生物学活性的重组体mPtCrustin2。
[1] Mu C, Zheng P, Zhao J, et al. A novel typeⅢcrustin (CrusEs2) identified from Chinese mitten crabEriocheirsinensis[J]. Fish Shellfish Immunol, 2011, 31 (1): 142-147.
[2] Smith VJ, Fernandes JM, Kemp GD, et al. Crustins: enigmatic WAP domain-containing antibacterial proteins from crustaceans[J]. Dev Comp Immunol, 2008, 32 (7): 758-772.
[3] Relf JM, Chisholm JR, Smith VJ, et al. Purification and characterization of a cysteine-rich 11.5 kDa antibacterial protein from the granular haemocytesof the shore crab,Carcinusmaenas[J]. Eur J Biochem, 1999, 264 (2): 350-357.
[4] Cui Z, Song C, Liu Y, et al. Crustins from eyestalk cDNA library of swimming crabPortunustrituberculatus: Molecular characterization, genomic organization and expression analysis[J]. Fish Shellfish Immunol, 2012, 33 (4): 937-945.
[6] 宋长丰,陶妍,赵冬梅,等. 斑点叉尾鮰铁调素成熟肽在毕赤酵母中的表达及其抑菌活性[J].农业生物技术学报,2015,23(3):380-387.
[7] 简思美,蔡斌斌,吴程,等.基因改造AiiA蛋白在毕赤酵母中的表达[J].福建师范大学学报(自然科学版),2015,31(6):55-63.
[8] 刘真英,李文利.密码子优化后的柞蚕溶菌酶在酵母中的表达及活性测定[J].微生物学通报,2016,43(2):292-300.
[9] 孙风敏,韩焱,李文利.基于密码子优化的蛋白酶K在毕赤酵母中的表达及分离纯化[J].微生物学通报,2014,41(11):2198-2207.
[10]杨刚刚,王泽,白海静,等.不同载体-宿主菌组合对重组蛋白表达量的影响[J].河南科技大学学报(自然科学版),2016,37(2):78-87.
[11]曹慕琛,徐健勇,罗立超,等.黑曲霉糖化酶基因的克隆及其在毕赤酵母X-33中的表达[J].安徽农业科学,2011,39(14):8226-8230.
Expression of Swimming Crab (Portunustrituberculatus) PtCrustin2 Mature Peptide inPichiapastoris
WANG Yan-hui, TAO Yan
(ShanghaiEngin.Res.Ctr.ofAqua-Prod’tProcess. &Preserv’n,Coll.ofFoodSci. &Technol.,ShanghaiOceanUni.,Shanghai201306)
Mainly distributing in crustaceans’ cells, crustins are small cysteine-rich antimicrobial peptides playing important roles in innate immune system of crustaceans. Crustins can be divided into several types according to their first structures. This paper takes PtCrustin2 mature peptide from swimming crab (Portunustrituberculatus) as researching target in order to realize recombinant DNA expression of the PtCrustin2 mature peptide inPichiapastoris, through the construction of aP.pastorisexpression system. First, total RNA was extracted from the gill of swimming crab. The cDNA encoding mPtCrustin2 mature peptide was amplified by RT-PCR, and inducedEcoRⅠandNotⅠrestriction endonuclease sites onto its 5′ and 3′ ends respectively. Then linked this target fragment onto the expression vector pPIC9K to construct the recombinant expression vector pPIC9K-mPtCrustin2, after it was e-transformed intoP.pastorisGS115 cell, and after screened and cultured with YPD medium containing different concentration of G418, high-copying yeast transformant was obtained and induced to express recombinant mPtCrustin2 through 0.5% methanol and immobilized metal affinity chromatography (IMAC) to isolate and obtained purified recombinant mPtCrustin2. Tricine-SDS-PAGE analysis indicated its molecular mass was 10.5 kDa. The inhibition experiment had proved that the recombinant mPtCrustin2 had a certain inhibition effect againstStaphylococcusaureusandPseudomonasaeruginosa. The present study for the first time realized the expression of recombinant DNA of mature peptide from swimming crab (P.trituberculatus) inP.pastoristhat lays foundation for further after-study.
swimming crab (Portunustrituberculatus); mPtCrustin2 mature peptide;Pichiapastoris; recombinant DNA expression
上海市教育委员会产学研项目(15CXY30);农业部都市农业(南方)重点实验室开放基金项目(UA201307)
王艳慧 女,硕士研究生。研究方向为水产生物分子生物学。E-mail:pacawang2013@126.com
* 通讯作者。女,教授,硕士生导师。研究方向为水产生物分子生物学。Tel: 021-61900384, E-mail:ytao@shou.edu.cn
2015-12-07;
2016-03-22
Q786
A
1005-7021(2016)05-0026-06
10.3969/j.issn.1005-7021.2016.05.005
