分子对接打分修正及在内皮脂肪酶选择性抑制剂筛选中的应用
2016-11-22罗棋耀王紫鋆金宏威刘振明张亮仁
罗棋耀 王紫鋆 金宏威 刘振明 张亮仁
(北京大学药学院,天然药物及仿生药物国家重点实验室,北京100191)
分子对接打分修正及在内皮脂肪酶选择性抑制剂筛选中的应用
罗棋耀王紫鋆金宏威刘振明*张亮仁*
(北京大学药学院,天然药物及仿生药物国家重点实验室,北京100191)
内皮脂肪酶(EL)是脂代谢调控甘油三酯脂酶家族的新成员,其功能主要为水解富含磷脂的高密度脂蛋白(HDL),对其进行选择性抑制而不影响其同源蛋白脂蛋白脂肪酶(LPL)能够提高血浆中HDLc的浓度水平,有利于预防及治疗动脉粥样硬化疾病。目前分子对接中的打分函数对大分子及大蛋白口袋具有偏向性,使得基于分子对接的虚拟筛选成功率普遍不高。本文中,我们将EL和LPL分别与Specs小分子库进行了分子对接,分析了对接打分与重原子数及接触面积的关系,发现对接打分与重原子数及接触面积之间有极高的相关性,即存在重原子数的叠加效应(重原子数越大,打分越好的趋势)。我们建立了基于重原子数和接触面积的EL、LPL对接打分标准曲线,利用此标准曲线进行对接打分的修正,并用已知抑制剂和生成的decoy分子作为验证集进行了验证。随后,我们应用此打分修正策略对传统中药库(TCMD)进行了基于分子对接的虚拟筛选,发现经过打分修正后的分子排名与重原子数之间的分布更为均衡,同时我们对EL打分较高、LPL打分较低且类药性较好的分子进行了结合模式的分析,为高活性高选择性EL抑制剂的发现奠定了基础。
内皮脂肪酶;脂蛋白脂肪酶;选择性抑制剂;分子对接;打分修正;标准曲线
1 Introduction
Endothelial lipase(EL),whose coding genes were identified by Hirata1and Jaye2et al.in 1999,is a new member of triglyceride lipase gene subfamily(TLGS)which plays a central role in the regulation of plasma lipid and lipoprotein metabolism3-6.TLGS is comprised of three high homologous lipases:lipoprotein lipase (LPL),hepatic lipase(HL),and endothelial lipase(EL).The homology of the sequences between ELand LPL,ELand HLwere 44%and 41%,respectively1.TLGS members were involved in the pathological process of hyperlipidemia and atherosclerosis through regulating the plasma cholesterol and triglyceride concentration7-10.Among these three lipases,LPL is mainly expressed in adipose and muscle tissues and mainly hydrolyzes the triglycerides of low-density lipoproteins(LDL)and chylomicrons11, whereas EL is synthesized by vascular endothelial cells,thyroid epithelial cells,and hepatocytes,exerting significant phospholipase activity on high-density lipoprotein(HDL)particles2,12.And HL has equal hydrolytic activity on triglycerides of LDL and phospholipids of HDL13.Elevated plasma low-density lipoprotein cholesterol(LDLc)levels are associated with increased risk of coronary artery disease(CAD)and atherosclerosis while plasma high-density lipoprotein cholesterol(HDLc)levels are negatively correlated with CAD7.HDL plays a main role in reverse cholesterol transport and is believed to be a CAD protective factor14.EL is the major negative regulator of plasma HDLc levels and involved in the pathological process of atherosclerosis through hydrolyzing HDL particles,which makes it a potent target for raising plasma HDLc in the treatment of atherosclerosis15-18.
The structures of EL,LPL,and HL share some common domains including HSPG binding domain and catalytic domain19. The variety and location of amino acids in catalytic domain are highly conserved,which have the similar characteristics as serine protease family20,21.The site-directed mutation demonstrates that these lipases all contain a GXSXG sequence and serine is the acylated center22-24.Due to the lack of EL,LPL,and HL crystal structures,our group has built these three lipase models using homology modeling method previously25.We compared these three lipases and found that ELhas a high similarity to LPLon the spatial structure,explaining why most known EL inhibitors(such as sulfonylfuran urea series and boronic acid series)also have the inhibition to LPL26-30.It would probably be an obstacle to design specific EL inhibitors based on EL structure.
From the current study,the plasma level of HDLc is negatively related to the function of EL15.EL mainly hydrolyzes HDL particles and releases free fatty acid,lysophosphatidylcholine,and ApoA2.ELcan also remodel HDLparticles and turn the large HDL particles to the smaller ones,reducing phospholipid and cholesteryl ester in them.What′s more,the loss of EL function caused by gene mutations significantly increased the plasma level of HDLc while it is reduced in EL overexpressing mice31.And HDL clearance is delayed in the EL knock-out mice experiment32. However,the function of LPL is opposite to that of EL.LPL mainly hydrolyzes very low-density lipoprotein(VLDL)which was a causative factor for atherosclerosis11.Overexpression or raising the activity of LPL could reduce the plasma triglyceride level and inhibit the development of atherosclerosis33.On the contrary,the loss of LPL function would lead to the siginificant elevation of VLDL and chylomicron.The biological activity data based on antibody indicated that selectively inhibiting EL could increase the plasma level of HDLc,and then reduce the risk of atherosclerosis34.Thus,developing specific EL inhibitors would be of great significance to the prevention and therapy for cardiovascular disease.
It is a great challenge to design specific EL inhibitors due to the high homology of EL and LPL as well as the low success rate of docking-based screening.Many contemporary scoring functions are influenced by the physical properties of docked molecules35. This bias can cause molecules with certain physical properties such as high molecular weight to incorrectly score better than others,which is a source of false positives in structure-based virtual screening36.In this article,we developed a statistical dock score correction strategy in docking-based virtual screening and applied it to the identification of specifc EL inhibitors(Fig.1). Firstly,we conducted the docking-based virtual screening of Specs database to EL and LPL,and we found the scoring bias phenomenon that the larger molecules and protein binding pockets usually obtained the lower binding energy.Then from the docking results of Specs database,we established the binding energy standard curves for EL and LPL based on heavy atom number (HA)and contact area(CA)to correct the dock energy score statistically,later validated the correction effects of these curves in the screening with a validation set.Furthermore,the traditional Chinese medicine database(TCMD)was screened by docking with the score correction strategy,and the dock ranks were compared before and after the correction to confirm the correction effectiveness of binding energy standard curves.Moreover,some compounds ranking at the top after the correction as well as some compounds with anti-hyperlipidemia activity which may be specific EL inhibitors exhibiting better binding affinity to EL than LPLwere analyzed to study the interaction mechanisms.The score correction strategy we proposed would be helpful to improve the hit rate in docking-based screening,and the molecules we found would be useful for experimental scientists in prioritizing drug candidates and studying the interaction mechanism of EL with itsspecific inhibitors.

Fig.1 Work flow of the score correction strategy in docking-based screening applied to specific ELinhibitors identification
2 Materials and methods
2.1Database preparation
2.1.1Standard virtual screening database
The commercially available Specs database was used here as a standard virtual screening database,which contains 200187 compounds.The molecules with more than 2 unclear chiral centers were removed.The database molecules were prepared by Ligprep37(Schrodinger Software)including ionization,generating isomers,and energy minimization.The molecules with more than 43 or less than 13 heavy atoms were filtered by Pipeline Pilot 7.538.Finally,192101 molecules were obtained for next docking procedure.
2.1.2Validation set
The validation set is composed of 22 anthranilic acid inhibitors of ELand 250 decoys.The anthranilic acid inhibitors were divided into 5 clusters by Discovery Studio 2.539and 1 inhibitor from each cluster was picked out.These 5 inhibitors were used to generate corresponding decoys with the rate 1:50.The criteria of molecular weight were set above 500 to meet our specific need validating the correctional effect on the scoring bias toward the large molecules.Totally 250 decoys were found from ZINC database by DecoyFinder40.The validation set consisting of 22 inhibitors and 250 decoys was prepared by Ligprep37(Schrodinger Software).
2.1.3External validation and application database
The TCMD built by Zhou′s laboratory was used here as an external validation and application database41,which contains totally 23033 compounds.The TCMD compounds with molecular weight greater than 800 were filtered out by Pipeline Pilot 7.538. Then the rest were prepared by Ligprep37(Schrodinger Software) as above,obtaining a total of 19680 compounds for virtual screening validation as well as application procedure.
2.2Establishing of binding energy standard curves
The Specs molecules were docked to the ELand LPLhomology models by AutoDock Vina42,respectively.Binding pockets were defined as the center of grid boxes for docking,and the size of each box was assigned as 2.2 nm×2.2 nm×2.2 nm.Docking calculations were performed using the default parameters implemented in AutoDock Vina42.Each molecule output 9 lowest binding energy conformations,and the best was selected as the binding energy of molecules.
The heavy atom(HA)number of Specs molecules was calculated by Pipeline Pilot 7.538and the docking complexes contact area(CA)between small molecules and macromolecule was calculated by an in-house python script.The average binding energy(AE)of each heavy atom number and contact area was calculated.The standard curves of binding energy and heavy atom number,binding energy and contact area were obtained using the exponential decay fitting method.And the exponential decay equation was used to describe the variation trend of the average binding energy with heavy atom number(HA)and contact area.
2.3Validation of the score correction effect
The validation set consisting of 22 anthranilic acid inhibitors of EL and 250 decoys were prepared and docked to EL and LPL by AutoDock Vina,respectively42.The binding energy standard curves were used to correct the dock energy score,eliminating the bias induced by heavy atom number and binding pocket properties.The original binding energy(EL_E,LPL_E)minus the corresponding average binding energy(EL_AE,LPL_AE)on the standard curve based on heavy atom number or contact area to obtain the correctional binding energy(EL_E′,LPL_E′).The correctional binding energy to ELwas used to evaluate the activity of molecules to EL,and the difference between EL binding energy and LPL binding energy(ΔE,ΔE′)was used to evaluate the se-lectivity of molecules to EL.The activity and selectivity evaluation of EL inhibitors and decoys before and after the correction were compared.The correctional binding energy and the difference between EL binding energy and LPL binding energy were calculated as follows:

2.4External validation and application with TCMD
The TCMD compounds were docked to EL and LPL by AutoDock Vina42.The default parameters were adopted as above. The binding energy standard curves were applied to correct the binding energy to EL and LPL.The original binding energy minus the corresponding binding energy on the standard curve based on heavy atom number or contact area to obtain the correctional binding energy.We used the correctional EL binding energy and the difference between ELbinding energy and LPLbinding energy to evaluate the activity and selectivity to EL,respectively.
As the score correction effects based on heavy atom number and contact area were similar,we only used the standard curve based on heavy atom number to correct the binding energy here. The compounds with ΔE′HAlower than-4.0 kJ·mol-1were retained,and then were ranked by EL_E′HA.Ten best compounds were selected as the potential specific EL inhibitors,and the top anti-hyperlipidemia compounds were selected as well.The druglikeness evaluation of the hits in TCMD was further implemented by the QED43module of Pipeline Pilot 7.5.
3 Results and discussion
3.1Binding site for specific EL inhibitors design
As mentioned above,the known EL inhibitors mostly lack selectivity to EL,inhibiting the homologous protein LPL as well. According to literature,these inhibitors are all covalent inhibitors forming covalent bond with the hydroxyl oxygen atom of catalytic serine26,28,29.Since the lipases family all have a catalytic triad and a certain degree of homology,the covalent EL inhibitors could also easily fit into the binding pocket of other lipases and bind with the catalytic serine,resulting in lacking selectivity to EL.

Fig.2 Comparison of ELbinding site and LPLbinding site
Thus,non-covalent inhibitors seem to be the better choice to bind with EL selectively.Fig.2 shows the binding site for specificEL inhibitors design.The binding pocket of EL resembles a deep, narrow hole(Fig.2(a))while that of LPL appears open,shallow, and quadrilateral(Fig.2(b)).The difference between EL and LPL binding pockets in shape provides a foundation to design specific EL inhibitors as the binding with EL and LPL needs a different spatial conformation.Furthermore,the binding site residues of EL and LPL were analyzed.As Fig.2(c)shows,the binding site residues of EL,such as TRP74,THR75,SER77,LEU210,HIS256, GLU257,TYR150,and ASP175,could serve as hydrogen bond acceptors to form hydrogen bonds with the hydrogen bond donors of ligands while ILE212,GLY213,LYS253,ASP229,and ARG206 may act as hydrogen bond donors.And there are some hydrophobic regions within the EL binding pocket.TRP55, THR56,SER132,SER240,PHE185,and VAL237 could serve as hydrogen bond acceptors while GLY159,ARG192,GLY191, ARG187,and ARG223 may act as hydrogen bond donors in the case of LPL,and its pocket is hydrophobic like that of EL(Fig.2 (d)).Though EL and LPL binding sites have some identical residues which may interact with ligands,the spatial positions and the distances of them are quite different.The binding pocket of LPL is large enough to fit the large molecules while the binding pocket of EL is suitable for the relatively small molecules.In consequence,the differences of EL and LPL binding sites provide the basis for docking-based virtual screening to find specific EL inhibitors.
3.2Docking-based virtual screening results of Specs database
The compounds of Specs database are docked to EL and LPL, respectively,obtaining the binding energy of compounds in EL and LPLbinding pockets.The distributions of binding energy with heavy atom number and contact area,which could be regarded as a background distribution,were analyzed.Fig.3 shows the binding energy distributions of Specs compounds,presenting the overall trend that binding energy decreased with the increasing heavy atom number and contact area.The larger heavy atom number and contact area usually result in the better energy score calculated by the docking program.This bias just like noise effect would make an adverse influence on docking-based screening,resulting in a lot of false positives which have a high molecular weight.And the binding energy to LPL would be gradually lower than that to EL with the increasing heavy atom number and contact area.As we can see,when the heavy atom number and contact area are at low value,the binding energy distribution of EL and LPL is similar. But when the heavy atom number and contact area are at high value,the binding energy to LPL is almost 4 kJ·mol-1lower than that to EL on the average,which may be caused by the difference of protein pocket properties.As the protein pocket properties may have noise effect on the calculated binding energy,the fact that the binding pocket of LPL is larger than that of EL results in a lower binding energy to LPL.Therefore,the dock binding energy scores to EL and LPL are not comparable.However,the bias can be eliminated by statistical correction.Based on the relationship between average binding energy and HAas well as CA,we could establish binding energy standard curves for EL and LPL to reflectthe average binding energy levels of molecules at each HA and CA.Then the binding energy could be corrected through deducting the corresponding binding energy on the standard curves. Then we could use the corrected binding energy to evaluate the binding affinity of molecules in an unbiased manner.
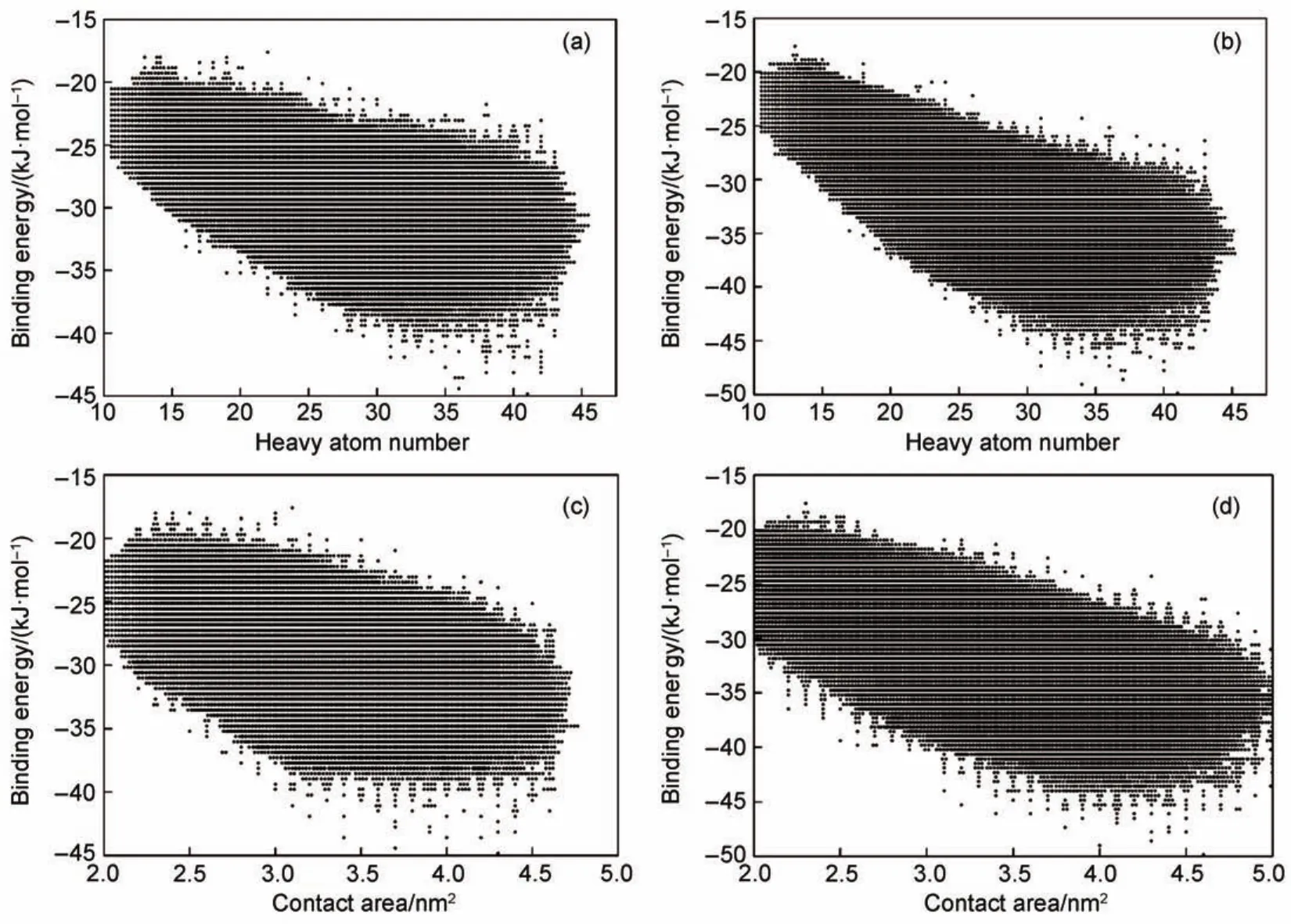
Fig.3 Docking energy score distribution of Specs compounds
3.3Virtual screening based binding energy standard curves
After analyzing the docking results of Specs compounds,it is known that there is a bias of dock energy scores toward the large molecules and binding pockets.To eliminate this bias,the binding energy standard curves were established using exponential decay fitting method based on the average binding energy at each HA and CA.The amount of compounds at each HA and CA is large enough to represent the overall binding energy levels.Thus the established standard curves would stand for the average binding energy of the molecules with different HA and CA.Fig.4 shows the EL and LPL binding energy standard curves for heavy atom number and contact area.As the heavy atom number and contact area increase,the binding energy decays exponentially and the difference between EL binding energy and LPL binding energy increases gradually.The standard curves decline fast at the beginning and would achieve a plateau when heavy atom number and contact area are at high values.It could be explained that when the molecules are larger than the binding pocket,the binding energy would not continue declining.And LPL binding energy is lower than ELbinding energy for LPLhas a larger binding pocket.
Heavy atom number and the average binding energy have the relationship described in Eq.(1)(EL)and Eq.(2)(LPL).

Fig.4 Binding energy standard curves
The HA-AEexponential decay fit curve of Specs docked to EL:

The HA-AE exponential decay fit curve of Specs docked to LPL:
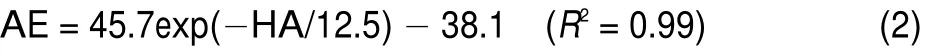
Contact area and the average energy score have the relationship described in Eqs.(3)(EL)and(4)(LPL).
The CA-AE exponential decay fit curve of Specs docked to EL:
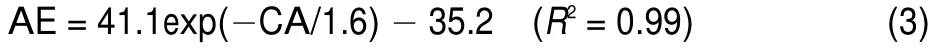
The CA-AE exponential decay fit curve of Specs docked to LPL:

The R2coefficient of the fitting curves are all as high as 0.99. This suggests that the exponential fitting curves can properly reflect the trend of the average binding energy.We speculate that it is not an individual case,which generally exists in the dockingbased virtual screening.And it has been proved in many other systems by our group.
3.4Score correction effect of binding energy standard curves
The validation set containing 22 EL inhibitors and 250 decoys was used to validate the effect of score correction strategy with binding energy standard curves.The decoys here are not real inactive molecules,just acting as the“background molecules”which has unclear activity to EL and LPL.Fig.5 shows the energy score distribution of the validation set.Most EL inhibitors are under the standard curve indicating that the binding energy is better than the average.The inhibitors above the standard curve based on heavy atom number are the same as that based on contact area.These inhibitors share some similar characteristics that they all have a highly flexible chain which may be hard to predict the binding pose and energy.As a result,their binding energy is predicted higher than other inhibitors in this case.As for the decoys,they are distributed on both sides of the curves evenly and some of them achieve a extremely low binding energy.Since the decoys mostly have a large molecular weight and heavy atom number,they are likely to obtain a lower binding energy and interfere the discovery of real inhibitors as false positives in dockingbased screening.In general,we use the line like the overall average line in Fig.5 to pick out the hits in the screening,which would probably result in a lot of large molecules with low binding energy.Alternatively,the binding energy standard curves could be used to pick out the active molecules more properly,reducing a lot of false positives with a large molecular weight(such as the points below the red line and above the standard curve in Fig.5)and retrieving the potential actives omitted in the general way(such as the points above the standard curve and below the red line in Fig.5).In this way,those molecules far under the curve may have a good binding affinity to the pocket and could be potent inhibitors of the target.Based on the original binding energy and correctional binding energy,the ROC curves were obtained to show the enrichment of EL inhibitors in the docking-based screening adopting the original score,the corrected score by HA,and the correctedscore by CA(Fig.6).We could see that the AUC values of both correctedscore(0.579 and 0.530)are far better than that of original score(0.374)and the correction by HA is a little better than that by CA,indicating the correctional strategy is effective to improve the true positive rate.Nevertheless,there is still some large molecules in front of the actives,which may be the disadvantage of this strategy that it can not correct the binding energy completely.On the other hand,it is still difficult for the docking programs to estimate the accurate binding energy presently.
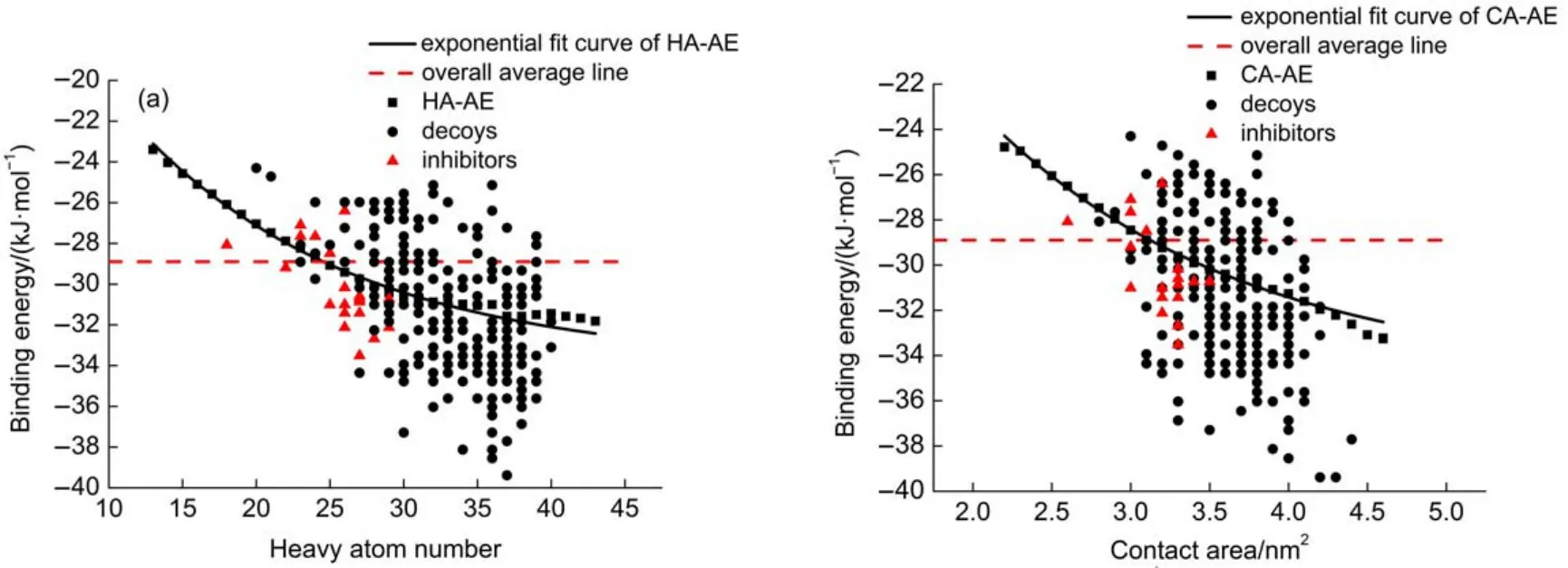
Fig.5 ELbinding energy distribution of validation set

Fig.6 ROC curves showing the enrichment of ELinhibitors in the docking-based screening adopting score correction by HA,score correction by CA,and original score
The difference between EL binding energy and LPL binding energy could be used to evaluate the selectivity of molecules to EL.Fig.7 shows the distribution of EL binding energy and LPL binding energy of validation set.As is shown,most inhibitors and decoys are under the diagonal line before correction indicating that LPL binding energy is lower than EL binding energy on the whole (Fig.7(a)),which is in agreement with the phenomenon in Fig.4. And the EL inhibitors which should have a lower binding energy to EL than LPL are distributed below the diagonal line,which is not consistent with the fact that these inhibitors are selective to EL.Apparently,the docking program has a bias toward LPL with a large pocket,resulting in that the binding energy to EL and LPL is not at the same level.Thus the binding energy of different binding pockets can not be compared equally because of the bias caused by the pocket properties.We could eliminate this bias by correcting the binding energy with the standard curves.After correction,we can see that the decoy is distributed on both sides of the diagonal line evenly and most EL inhibitors are above the diagonal line(Fig.7(b),Fig.7(c))which is consistent with the selectivity of EL inhibitors.Therefore,the difference between the correctional EL binding energy and correctional LPL binding energy could be used to evaluate the selectivity of molecules.The smaller the value of ΔE′is,the more selective to ELthe molecules would be.In other words,the molecules far above the diagonalmay be highly selective to EL.It would be helpful for us to pick out the potential specific EL inhibitors in this way.

Fig.7 Plot of the LPLbinding energy(y-axis)versus the ELbinding energy(x-axis)of validation set

Fig.8 Comparison of the distribution of score rank adopting differenct strategies with heavy atom number
To some extent,the binding energy standard curves help to eliminate the noise effect caused by heavy atom number and protein binding pocket properties.But it can not correct the binding energy completely.Apart from the heavy atom number, contact area,and pocket properties,there are some other factors that may influence the accuracy of binding energy.More or less, the score correctional strategy here helps to improve the true positive rate and identify the selective molecules on the existing docking-based screening.
3.5External validation and application with TCMD 3.5.1Virtual screening results of TCMD
Natural products have a lot of advantages such asdiverse structures,various bioactivities,less side effects,and a rich source, help to play an important role in the development of new drugs and lead compounds44.TCMD was built by Zhou′s laboratory, which contains more than twenty thousand natural products.As one of its characteristics,the molecular weight of TCMD compounds is of great difference.There is a high possibility that virtual screening of TCMD would result in a lot of false positives, especially for the proteins which have a small binding pocket such as EL.Thus the correcting method we develop could be used in the screening of TCMD to reduce false positives.In addition,most of the TCMD compounds have various bioactivities,and among them,some show anti-hyperlipidemia and anti-atherosclerosis activities,which may be the inhibitors of EL.Therefore,TCMD is used as the validation database to validate the effect of the score correction strategy we developed,and find potent specific EL inhibitors as well.

Fig.9 Plot of ΔE′HA(y-axis)versus EL_E′HA(x-axis)
The docking procedure was implemented and the binding energy score to EL and LPL of all 19660 compounds in TCMD was obtained.Then we used the binding energy standard curves established before to correct the binding energy which could eliminate the bias introduced by heavy atom number as well as protein pocket properties.We compared the rank of compounds based on EL binding energy before and after the correction.Fig.8 shows the distribution of the EL binding energy rank with heavy atom number of TCMD compounds.We can see that most of the original ranks in front are distributed on the region of molecules with large heavy atom number(Fig.8(a)).And the molecules with heavy atom number smaller than 20 mostly rank behind.The average rank of compounds at each heavy atom number decreased along with the increasing heavy atom number,which is consistent with that in the Specs database the molecules with larger heavy atom number usually obtain the lower binding energy.The top 20compounds almost all have a heavy atom number greater than 35 (Fig.8(d)).And it results in most of the molecules we captured have a large molecular weight which are likely to be false positives.It could be a reason why the success rate of docking-based virtual screening is still low.After the correction based on heavy atom number and contact area,the distribution of EL binding energy rank with heavy atom number is more balanced(Fig.8(b, c)).The molecules ranking in front mostly focus on the heavy atom number from 10 to 30 indicating that the molecules in this range are more proper for the binding with EL binding pocket. And most molecules with heavy atom number larger than 45 rank behind for these molecules are too large to fit the pocket.The heavy atom number of the top 20 compounds ranges from 20 to 55,mainly focusing on the range 25 to 40,which is more promising to be potential hits(Fig.8(e,f)).

Table 1 The best 10 compounds found via virtual screeening from TCMD
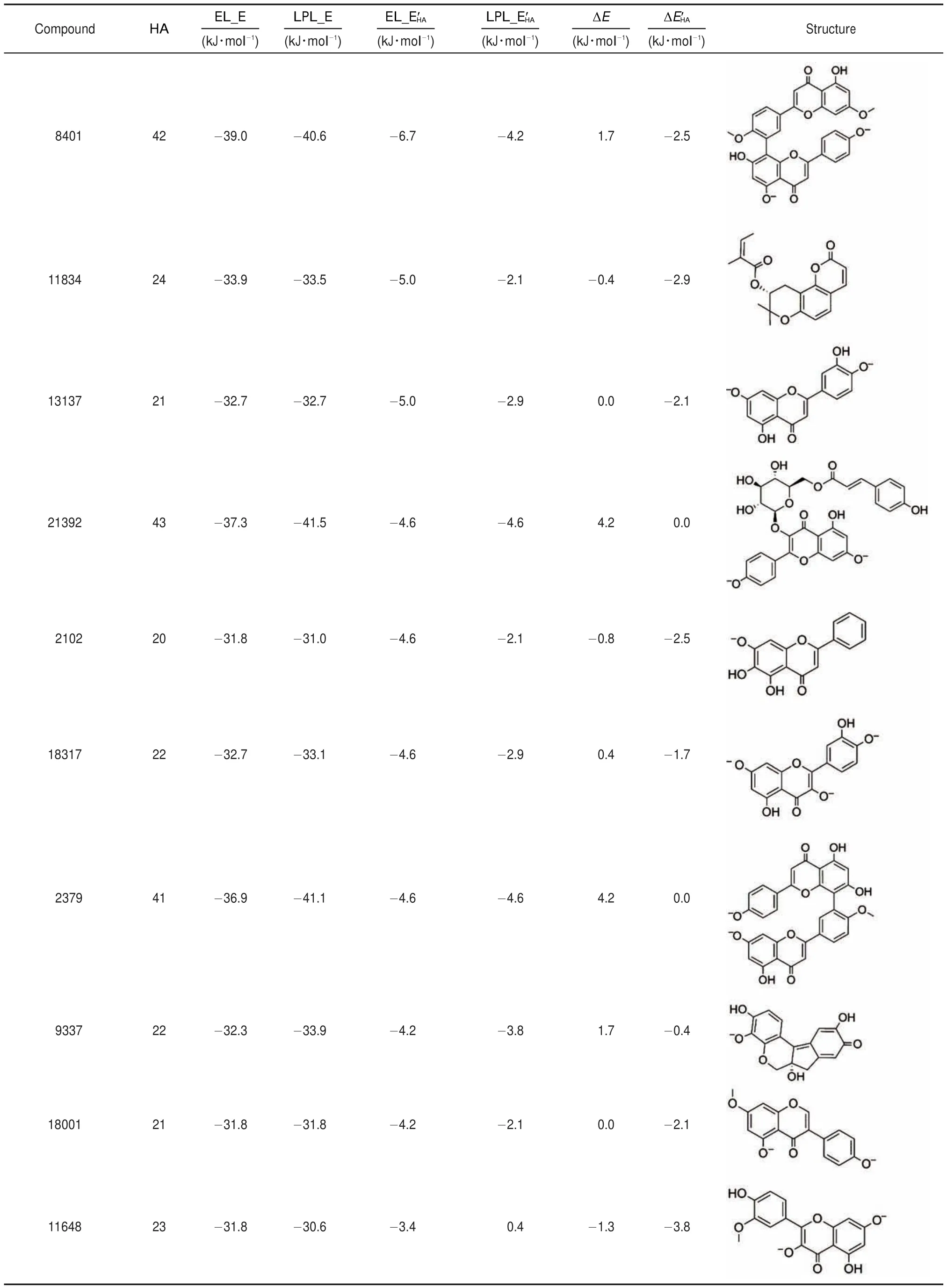
Table 2 The top 10 anti-hyperlipidemia compounds in TCMD
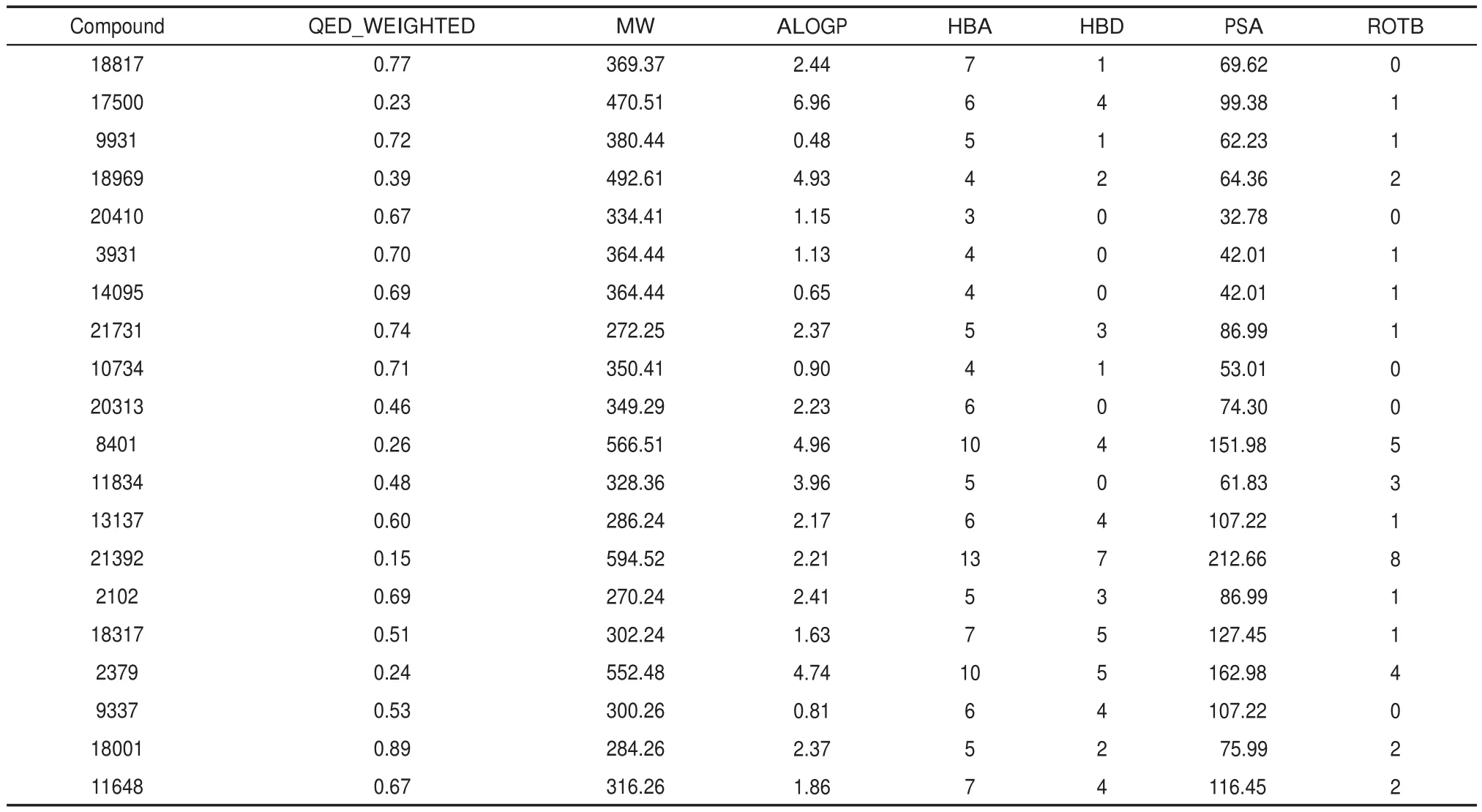
Table 3 The drug likeness and main properties of hits in TCMD
As we aim at finding the EL specific inhibitors,the correctional EL binding energy(EL_E′HA)and the difference between correctional EL binding energy and correctional LPL binding energy (ΔE′HA)were combined to pick the potential molecules(Fig.9).The points inside the circle with both low EL_E′HAand low ΔE′HArepresents the potential specific EL inhibitors,which would bind more tightly to EL than LPL.For the correctional effect based on heavy atom number and contact area is similar,here we only use the correctional binding energy based on heavy atom number to pick the TCMD compounds.Compounds with ΔE′HAsmaller than-4.0 kJ·mol-1were retained to rank by EL_E′HA.The structures of the top 10 compounds and their binding energy score are listed in Table 1,and the top 10 anti-hyperlipidemia compounds are listed in Table 2.
3.5.2Analysis of hits in TCMD
The drug likeness and main properties of hits in TCMD are shown in Table 3.We can see that most hits are drug-like with quantitative estimate of drug-likeness(QED)(the drug likeness is better when QED is closer to 1)more than 0.5,except for individual hits.It demonstrates that these hits have the potential to develop as anti-atherosclerosis drugs.Furthermore,the binding modes of TCMD compounds 18817 and 13137 with EL binding site were analyzed.These two compounds both achieve excellent docking and drug-likeness evaluation.They have similar interactions with the residues of EL binding site.Compound 18817 is a molecule with six continuous rings,just fitting into the deep hole.And the oxygen atoms of the tetrahydrofuran rings at the ends form hydrogen bonds with the hydroxyl groups of THR75 and TYR150(Fig.10(a,c)).Because of the strong hydrophobicity of the rings,it could interact with LEU210,SER211,ILE212,and HIS256 by hydrophobic contact.Compound 13137 was a molecule with anti-hyperlipidemia activity which might target EL.Its interactional mode is similar with compound 18817,forming hydrogen bonds with SER77,TYR150,HIS256 and hydrophobic contact with THR75,MET76,SER211,ILE212,and HIS256 (Fig.10(b,d)).The binding conformations of these compounds are fitting well with the spatial shape of EL binding pocket and their interactions with the binding site residues through hydrogen bonds and hydrophobic contacts are strong enough to bind with EL. Simultaneously,their binding energy score to LPL is far worse than EL,ensuring their binding selectivity to EL.These com-pounds will be useful for experimental scientists in prioritizing drug candidates and studying the interaction mechanism.

Fig.10 Binding modes and interactions of ELwith its potential inhibitors
4 Conclusions
In this work,we have developed a score correction strategy in docking-based virtual screening and have found some potential EL specific inhibitors applying this strategy.Firstly in the docking screening of Specs database against EL and LPL,we found the scoring bias phenomenon that the larger molecules and protein binding pockets usually obtained the lower binding energy which was probably induced by heavy atom number of compounds as well as the pocket properties of proteins,and then established the binding energy standard curves for EL and LPL based on heavy atom number and contact area,which could be used to correct the binding energy score of docking.The correction effect of the binding energy standard curves was validated with a validation set consisiting of EL inhibitors and decoys with large molecular weight.Furthermore,the TCMD was screened by docking with the score correction strategy,which could act as a further validation of the correction effect,showing that after the correction the compounds ranked in front were more properly than before. Besides,some compounds ranked in front including some antihyperlipidemia compounds with better binding affinity to EL than LPL were selected and their interation mechanisms were studied, which would probably be specific ELinhibitors.These compounds would be useful for experimental scientists in prioritizing drug candidates and would provide groundwork for potential therapies of hyperlipidemia and atherosclerosis.Moreover,it can be assumed that the score correction approach we put forward can be profitably applied also for other targets to find specific hits.
References
(1) Hirata,K.;Dichek,H.L.;Cioffi,J.A.;Choi,S.Y.;Leeper,N. J.;Quintana,L.;Kronmal,G.S.;Cooper,A.D.;Quertermous,T.J.Biol.Chem.1999,274(20),14170.doi:10.1074/ jbc.274.20.14170
(2) Jaye,M.;Lynch,K.J.;Krawiec,J.;Marchadier,D.;Maugeais, C.;Doan,K.;South,V.;Amin,D.;Perrone,M.;Rader,D.J. Nat.Genet.1999,21(4),424.doi:10.1038/7766
(3) Wong,H.;Schotz,M.C.J.Lipid.Res.2002,43(7),993. doi:10.1194/jlr.R200007-JLR200
(4) Fan,J.;Wang,J.;Bensadoun,A.;Lauer,S.J.;Dang,Q.; Mahley,R.W.;Taylor,J.M.Proc.Natl.Acad.Sci.U.S.A. 1994,91(18),8724.doi:10.1073/pnas.91.18.8724
(5) Zhang,J.;Yu,Y.;Nakamura,K.;Koike,T.;Waqar,A.B.; Zhang,X.;Liu,E.;Nishijima,K.;Kitajima,S.;Shiomi,M.;Qi, Z.;Yu,J.;Graham,M.J.;Crooke,R.M.;Ishida,T.;Hirata,K.; Hurt-Camejo,E.;Chen,Y.E.;Fan,J.J.Atheroscler.Thromb. 2012,19(3),213.doi:10.5551/jat.11148
(6) Goldberg,I.J.J.Lipid.Res.1996,37(4),693.
(7) Ichikawa,T.;Liang,J.;Kitajima,S.;Koike,T.;Wang,X.;Sun, H.;Morimoto,M.;Shikama,H.;Watanabe,T.;Yamada,N.;Fan, J.Atherosclerosis 2005,179(1),87.doi:10.1016/j. atherosclerosis.2004.10.044
(8) Nong,Z.;Gonzalez-Navarro,H.;Amar,M.;Freeman,L.; Knapper,C.;Neufeld,E.B.;Paigen,B.J.;Hoyt,R.F.;Fruchart-Najib,J.;Santamarina-Fojo,S.J.Clin.Invest.2003,112(3), 367.doi:10.1172/JCI200316484
(9) Tsutsumi,K.;Inoue,Y.;Shima,A.;Iwasaki,K.;Kawamura,M.; Murase,T.J.Clin.Invest.1993,92(1),411.doi:10.1172/ JCI116582
(10) Dalan,A.B.;Toptas,B.;Bugra,Z.;Polat,N.;Yilmaz-Aydogan, H.;Cimen,A.;Isbir,T.Mol.Biol.Rep.2013,40(8),5143. doi:10.1007/s11033-013-2615-2
(11) Merkel,M.;Eckel,R.H.;Goldberg,I.J.J.Lipid.Res.2002,43 (12),1997.doi:10.1194/jlr.R200015-JLR200
(12) McCoy,M.G.;Sun,G.S.;Marchadier,D.;Maugeais,C.;Glick, J.M.;Rader,D.J.J.Lipid.Res.2002,43(6),921.
(13) Connelly,P.W.Clin.Chim.Acta 1999,286(1),243. doi:10.1016/S0009-8981(99)00105-9
(14) Martinez,L.O.;Jacquet,S.;Terce,F.;Collet,X.;Perret,B.; Barbaras,R.Cell Mol.Life Sci.2004,61(18),2343. doi:10.1007/s00018-004-4087-y
(15) Ishida,T.;Choi,S.;Kundu,R.K.;Hirata,K.;Rubin,E.M.; Cooper,A.D.;Quertermous,T.J.Clin.Invest.2003,111(3), 347.doi:10.1172/JCI200316306
(16) Tatematsu,S.;Francis,S.A.;Natarajan,P.;Rader,D.J.; Saghatelian,A.;Brown,J.D.;Michel,T.;Plutzky,J. Arterioscler.Thromb.Vasc.Biol.2013,33(8),1788. doi:10.1161/ATVBAHA.113.301300
(17)Annema,W.;Tietge,U.J.Curr.Atheroscler.Rep.2011,13(3), 257.doi:10.1007/s11883-011-0175-2
(18) Degoma,E.M.;Rader,D.J.Nat.Rev.Cardiol.2011,8(5),266. doi:10.1038/nrcardio.2010.200
(19) Nardini,M.;Dijkstra,B.W.Curr.Opin.Struct.Biol.1999,9 (6),732.doi:10.1016/S0959-440X(99)00037-8
(21) Derewenda,Z.S.;Derewenda,U.Biochem.Cell Biol.1991,69 (12),842.doi:10.1139/o91-125
(22) Emmerich,J.;Beg,O.U.;Peterson,J.;Previato,L.;Brunzell,J. D.;Brewer,H.B.,Jr.;Santamarina-Fojo,S.J.Biol.Chem.1992, 267(6),4161.
(23) Davis,R.C.;Stahnke,G.;Wong,H.;Doolittle,M.H.;Ameis, D.;Will,H.;Schotz,M.C.J.Biol.Chem.1990,265(11),6291. (24) Lowe,M.E.J.Biol.Chem.1992,267(24),17069.
(25) Wang,Z.;Li,S.;Sun,L.;Fan,J.;Liu,Z.PLoS One 2013,8(8), e72146.doi:10.1371/journal.pone.0072146
(26) Greco,M.N.;Connelly,M.A.;Leo,G.C.;Olson,M.W.; Powell,E.;Huang,Z.;Hawkins,M.;Smith,C.;Schalk-Hihi,C.; Darrow,A.L.;Xin,H.;Lang,W.;Damiano,B.P.;Hlasta,D.J. Bioorg.Med.Chem.Lett.2013,23(9),2595.doi:10.1016/j. bmcl.2013.02.113
(27) Sun,S.;Dean,R.;Jia,Q.;Zenova,A.;Zhong,J.;Grayson,C.; Xie,C.;Lindgren,A.;Samra,P.;Sojo,L.;van Heek,M.;Lin, L.;Percival,D.;Fu,J.M.;Winther,M.D.;Zhang,Z.Bioorg. Med.Chem.2013,21(24),7724.doi:10.1016/j. bmc.2013.10.023
(28) Goodman,K.B.;Bury,M.J.;Cheung,M.;Cichy-Knight,M. A.;Dowdell,S.E.;Dunn,A.K.;Lee,D.;Lieby,J.A.;Moore, M.L.;Scherzer,D.A.;Sha,D.;Suarez,D.P.;Murphy,D.J.; Harpel,M.R.;Manas,E.S.;McNulty,D.E.;Annan,R.S.; Matico,R.E.;Schwartz,B.K.;Trill,J.J.;Sweitzer,T.D.; Wang,D.Y.;Keller,P.M.;Krawiec,J.A.;Jaye,M.C.Bioorg. Med.Chem.Lett.2009,19(1),27.doi:10.1016/j. bmcl.2008.11.033
(29) O'Connell,D.P.;LeBlanc,D.F.;Cromley,D.;Billheimer,J.; Rader,D.J.;Bachovchin,W.W.Bioorg.Med.Chem.Lett.2012, 22(3),1397.doi:10.1016/j.bmcl.2011.12.043
(30)Abdel-Magid,A.F.ACS Med.Chem.Lett.2013,4(11),1016. doi:10.1021/ml400361q
(31)Jensen,M.K.;Rimm,E.B.;Mukamal,K.J.;Edmondson,A. C.;Rader,D.J.;Vogel,U.;Tjonneland,A.;Sorensen,T.I.; Schmidt,E.B.;Overvad,K.Eur.Heart J.2009,30(13),1584. doi:10.1093/eurheart/ehp145
(32) Tanaka,H.;Ishida,T.;Johnston,T.P.;Yasuda,T.;Ueyama,T.; Kojima,Y.;Kundu,R.K.;Quertermous,T.;Ishikawa,Y.; Hirata,K.J.Atheroscler.Thromb.2009,16(4),327. doi:10.5551/jat.No844
(33) Fan,J.;Unoki,H.;Kojima,N.;Sun,H.;Shimoyamada,H.; Deng,H.;Okazaki,M.;Shikama,H.;Yamada,N.;Watanabe,T. J.Biol.Chem.2001,276(43),40071.doi:10.1074/jbc. M105456200
(34) Jin,W.;Millar,J.S.;Broedl,U.;Glick,J.M.;Rader,D.J.J. Clin.Invest.2003,111(3),357.doi:10.1172/JCI200316146
(35) Wallach,I.;Jaitly,N.;Nguyen,K.;Schapira,M.;Lilien,R.J.Chem.Inf.Model.2011,51(8),1817.doi:10.1021/ci200175h
(36) Jacobsson,M.;Karlen,A.J.Chem.Inf.Model.2006,46(3), 1334.doi:10.1021/ci050407t
(37) LigPrep2.6;Schrödinger,LLC:New York,2013.
(38) Pipeline Pilot 7.5;Accelrys Software Inc.:San Diego,CA, USA.http://accelrys.com(accessed January 25,2015).
(39) Discovery Studio 2.5;Accelrys Software Inc.:San Diego,CA, USA.http://accelrys.com(accessed January 25,2015).
(40) Cereto-Massague,A.;Guasch,L.;Valls,C.;Mulero,M.; Pujadas,G.;Garcia-Vallve,S.Bioinformatics 2012,28(12), 1661.doi:10.1093/bioinformatics/bts249
(41) He,M.;Yan,X.;Zhou,J.;Xie,G.J.Chem.Inf.Model.2001,41 (2),273.doi:10.1021/ci0003101
(42) Trott,O.;Olson,A.J.J.Comput.Chem.2010,31(2),455. doi:10.1002/jcc.21334
(43) Bicherton,G.R.;Paolini,G.V.;Besnard,J.;Muresan,S.; Hopkins,A.L.Nat.Chem.2012,4(2),90.doi:10.1038/ nchem.1243
(44) Newman,D.J.;Cragg,G.M.J.Nat.Prod.2007,70(3),461. doi:10.1021/np068054v
Improved Docking-Based Virtual Screening Using the Score Correction Strategy for Specific Endothelial Lipase Inhibitors Identification
LUO Qi-YaoWANG Zi-YunJIN Hong-WeiLIU Zhen-Ming*ZHANG Liang-Ren*
(State Key Laboratory of Natural and Biomimetic Drugs,School of Pharmaceutical Sciences, Peking University,Beijing 100191,P.R.China)
Endothelial lipase(EL)has been implicated in high-density lipoprotein(HDL)metabolism and the pathogenetic progress of atherosclerosis,so its specific inhibitors are expected to be useful for the treatment of cardiovascular disease.In addition to the high homology of EL with other lipases such as lipoprotein lipase (LPL),the scoring bias of current docking programs toward large molecules and large protein-binding pockets also makes it difficult to find specific EL inhibitors by docking-based virtual screening.Herein,we conducted docking-based virtual screening of the Specs database for EL and LPL firstly,and we found the scoring bias phenomenon.From the docking results of the Specs database,we established standard curves for the binding energies of EL and LPL based on heavy atom number and contact area to correct the dock energy score statistically.We then validated the correctional effects of these curves in the screening of a validation set. Furthermore,the traditional Chinese medicine database(TCMD)was screened by docking using the score correction strategy.The dock ranks before and after correction were compared to confirm the screening effectiveness.Moreover,some compounds exhibiting better affinity for EL than LPL after correction as well as some compounds with antihyperlipidemic activity that may be specific EL inhibitors were analyzed to study their interaction mechanisms.The developed score correction strategy should be helpful to improve the hit rate indocking-based virtual screening.The molecules we identified should be useful for experimental scientists to prioritize drug candidates and provide groundwork for potential therapies of hyperlipidemia and atherosclerosis.
April 21,2016;Revised:June 20,2016;Published online:June 20,2016.
s.LIU Zhen-Ming,Email:zmliu@bjmu.edu.cn;Tel:+86-10-82805514.ZHANG Liang-Ren,Email:liangren@bjmu.edu.cn; Tel:+86-10-82802567.
Endothelial lipase;Lipoprotein lipase;Specific inhibitor;Molecular docking;Score correction;Standard curve
O641
10.3866/PKU.WHXB201606202
The project was supported by the National Natural Science Foundation of China(21572010,21272017).
国家自然科学基金(21572010,21272017)资助项目©Editorial office ofActa Physico-Chimica Sinica
(20) Brenner,S.Nature 1988,334(6182),528.10.1038/ 334528a0
