Fe、Al纳米粒子熔化过程结构转变的分子动力学研究
2020-03-26张宇赵鹤平
张宇赵鹤平
(1.吉首大学 物理与机电工程学院,吉首 416000;2.湖南工程学院 计算科学与电子学院,湘潭 411101)
0 引言
金属纳米粒子作为一种催化材料,在催化功能和特性方面表现出优异的性能[1-3],实验研究表明金属纳米粒子与块体金属材料在物理性能和化学性能之间有着明显的不同[1-3],因此其熔化行为和块体金属也会有着显著的差异.Fe、Al在生活中是一种常见金属且应用广泛,对其纳米粒子的熔化特性进行研究有着重要意义.实验与理论研究都表明,金属纳米粒子的熔点、熔化行为与其尺寸有密切关系[4],而小尺度纳米粒子的熔化行为更加丰富[4-5],其中可能涉及到结构的转变或固液共存状态.I Hamid, et al.[4]在其工作中就提到了当原子数为13和55时,截角八面体(Oh)结构的Al纳米粒子在低温不能稳定存在,他们还比较了较大尺寸Oh和ICO结构的热力学稳定性,更细致地研究金属纳米粒子熔化行为与粒子尺寸、粒子构型的关系.在以往的研究中,大多数采用的是经典的嵌入原子势,而没有引入能量修正项.
本文选择Fe、Al两种金属,模拟了粒子数从55到561的几种构型的纳米粒子的熔化行为,应用分子动力学方法结合势能、共近邻与径向分布函数等技术分析方法,讨论了熔化过程中的结构转变.采用了改进型的嵌入原子势结合分子动力学方法,模拟了Fe、Al金属纳米粒子熔化行为,纳米粒子的升温每次只有10 K,比以往的研究温度上升的更细.比较了在不同尺寸和构型下Fe、Al金属纳米热力学稳定性.
1 计算模型
本文采用了嵌入原子势模型,并结合分子动力学模拟了Fe-ICO(原子数分别为147、309、561)、Al-ICO(原子数分别为147、309、561)、Al-Dh(原子数分别为55、147、309、561)和Al-CO(原子数分别为55、147、309、561)的熔化过程中的热力学行为.该势已经成功地被用于研究金属以及合金的块体、表面和团簇[6-9].在嵌入原子势模型中,系统的总能量由下式给出[10].
(1)
其中Ei是这个系统中第i个原子的总能.
(2)
关于该势能更加详细的表达,请参考文献[10].
在图1中分别绘制了原子数为561的三种结构示意图,对于一个具有l层的理想ICO结构的纳米粒子,Dh结构和CO结构其壳层数与粒子数的关系与ICO结构一样,其壳层数与粒子数的关系如下所示[5].
(3)

图1 (a)表示原子数为561的ICO结构的纳米粒子;
(b)表示原子数为561的Dh结构的纳米粒子;
(c)图表示原子数为561的CO结构的纳米粒子
2 结果与讨论
2.1 Fe-ICO纳米粒子
为了探究完整的Fe-ICO结构的熔化行为,我们利用分子动力学程序对整个纳米粒子进行连续加热,模拟了原子数分别为147、309、561的熔化过程.我们分别提取系统的势能,用共近邻方法(CNA)进行分析比较.图2分别绘制了Fe-ICO的势能随温度的依赖关系,从图中可以看到随着温度的升高,粒子的总体势能呈现线性的增加,粒子的熔点随着原子数的增加而增加.原子数分别为147、309、561的纳米粒子在温度分别为1080 K、1180 K、1280 K处势能有一个跃迁,由此我们可以判断,粒子在该温度下经历了从固态向液态的转变,也就是熔化.从图3中可以看到在原子数为147个时,其中(4,2,1)键的比例从1080 K加热到1090 K时发生骤降,从0.32降低到0.08,综合上述几个物理量的变化,我们能判断出这几个纳米粒子的熔点.原子数分别为309、561的Fe-ICO结构的纳米粒子也能看到类似的结果,在这里便不再一一赘述.显然这三种判断方法显示的结果有着很好的一致性,都表明了纳米粒子在此温度熔化了.

图2 理想的Fe-ICO结构(原子数分别为147、309、561)的势能随温度的变化关系图
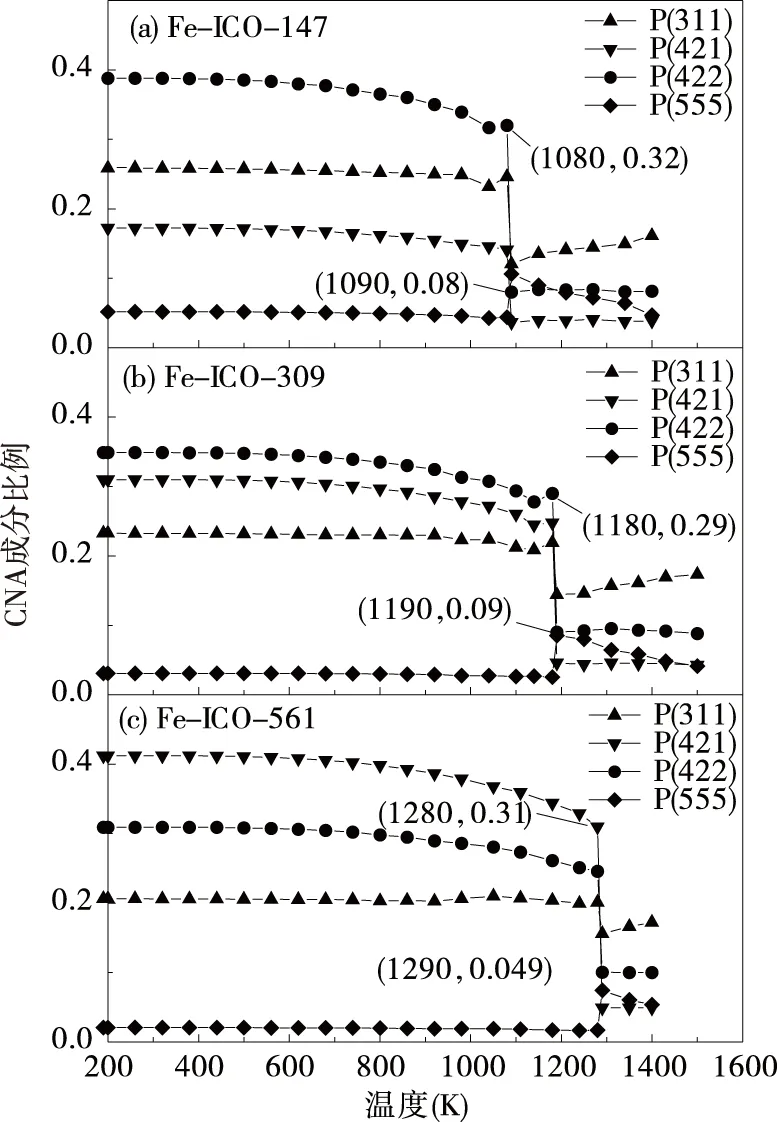
图3 原子数分别为147、309、561的理想的Fe-ICO结构CNA各成分比例随温度的变化关系图
2.2 Al-ICO纳米粒子
为进一步理解纳米粒子的熔化过程,图4绘制了模拟过程中原子数量为147、309、561的Al-ICO结构的纳米粒子势能随温度的变化关系.其结果和Fe-ICO结构的模拟结果类似,较低温度时势能随温度的升高而线性增加,在到达某一个温度势能有一个明显的跃迁.原子数量为147、309、561的纳米粒子对应的势能曲线发生跃迁的温度分别为560 K、650 K、700 K.在这两个过渡临界温度前后势能基本上呈现出线性增加的趋势.这与很多其他作者类似的实验结果[4-5,14-15]是吻合的.
为了让我们的模拟结果更加具有说服性,我们也应用CNA方法分析了Al-ICO(原子数量分别为147、309、561)纳米粒子熔化过程,其结果与对应的势能分析结果有很好的一致性.
在图4和图5中,随着温度升高,其曲线的波动情况越来越剧烈,这是由于温度升高,原子在平衡位置的波动程度越来越剧烈导致的,甚至会有内外层原子之间的扩散发生.这在之前的一些研究[12,14]有过类似的报导.结果表明,纳米粒子会有表面预熔现象发生,从壳外逐渐熔化扩散到壳内熔化,最后直到整个纳米粒子全部熔化.

图4 理想的Al-ICO结构的势能随温度的变化关系图

图5 理想的Al-ICO结构CNA各成分比例随温度的变化图
2.3 Al-Dh纳米粒子
这一节,原子数为55、147、309、561的Al-Dh纳米粒子的结果.图6给出了原子数量为55、147、309、561的Al-Dh纳米粒子势能随温度的关系曲线.从图6(a)、图6(b)中可以发现,在持续升温的过程中,纳米粒子势能有一个下降的过程(对应的温度分别为90 K和290 K),而在图6(c)、图6(d)两图中并没有发现这种现象.为了探究这一现象的原因,采用CNA方法分析Al-Dh纳米粒子在持续加热过程中的结构变化过程.图7中分别给出原子数为55、147、309、561的Al-Dh纳米粒子的CNA曲线.从图7(a)可以发现在温度从90 K升高到100 K时,(4,2,2)、(3,1,1)、(5,5,5)指数的比例有一个急剧的增加,之后又变得比较稳定.(4,2,2)键比例的增加表明hcp原子增多,(3,1,1)键比例增加表明(1,1,1)面的原子数量变多,(5,5,5)键比例的增加表明局部处在五重对称轴的原子数增加.当温度继续从340 K上升到350 K时,(4,2,2)键比例却开始急剧的下降,然后再继续增加温度在370 K至390 K之间,(4,2,2) 键比例又再一次发生急剧的下降,说明纳米粒子已经全部熔化.在图7(b)中也可以发现类似的结果,即在升温熔化过程中,纳米粒子的结构会先向ICO结构转变.然而在图7(c)、图7(d)两图中并没有这样类似的现象发生,即Al-Dh纳米粒子所表现出来的一类新现象.

图6 (a)、(b)、(c)、(d)分别表示的是理想的Al-Dh结构(原子数分别为55、147、309、561)的势能随温度的变化关系
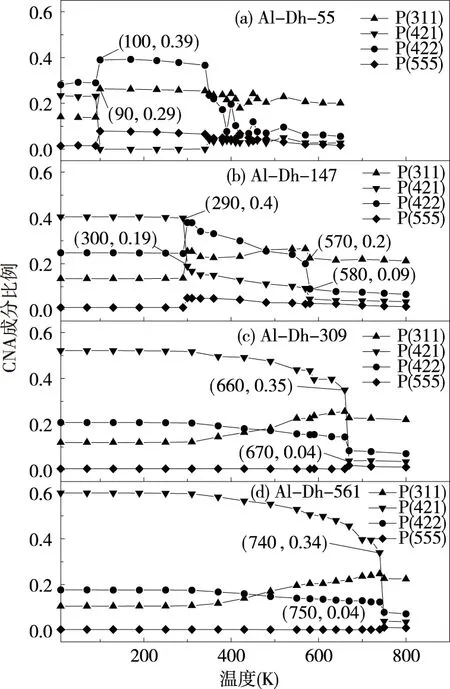
图7 原子数分别为55、147、309、561的理想的Al-Dh结构CNA各成分比例随温度的变化关系
为了更加细致地描述上述的过程,图8中绘制了在不同温度下原子数分别为55和147的Al-Dh纳米粒子的瞬时形状,即原子坐标示意图.从图8中看到,原子数为55、147的Al-Dh在温度逐渐升高时,由原来的Dh结构转变为了ICO结构.原子数为55、147的Al-Dh的纳米粒子,温度升高的时候会发生结构的转变,是因为在原子数比较少的时候ICO结构比Dh结构更加稳定,当温度升高到一定程度的时候,这个时候粒子就会具有足够的能量使自己的结构发生重排,形成能量更小的结构,以前的研究工作[5,12,16]也报导了这种从Dh向ICO结构转变的现象.

图8 在持续加热过程中不同的温度下完整的Al-Dh结构的纳米粒子(原子数量分别为55、147)的瞬时形状
2.4 Al-CO纳米粒子
这一部分研究原子数量为147、309、561完整的Al-CO纳米粒子的熔化过程.图9(a)、图9(b)、图9(c)表示数量为147、309、561的Al-CO纳米粒子在连续加热过程中势能随温度的变化曲线.在这三个图中可以看到,除熔化转变温度外,其对应的势能分别随温度升高而线性增加.然后温度增加到熔点附近时候,势能有一个跃迁的过程,说明整个粒子在该温度下发生了熔化,成为了液态.
同样也绘制了Al-CO结构的CNA图,对于原子数量为147的Al-CO结构的纳米粒子,发现其CNA图明显不同于另外两种数量的纳米粒子.在图10(a)图中,(421)键比例变化剧烈,在510 K时有一个比较大的变化,然后在势能图中发现并没有很明显的能量变化.为了更加直观的看到这一过程,图11中绘制了原子数为147的Al-CO在不同温度下纳米粒子的瞬时形状,发现在520 K时其粒子的构型已经发生了转变,从最初的Dh结构转变成了ICO结构.同之前原子数为147的Al-Dh一样,在熔化之前都有一个结构的转变过程,然而对于原子数为309、561的Al-CO结构在熔化之前没有发生结构的转变,与Al-Dh结构的情形一样.

图9 (a)、(b)、(c)分别表示的是理想的Al-CO结构(原子数分别为147、309、561)的势能随温度的变化关系

图10 原子数分别为147、309、561的理想的Al-CO结构CNA各成分比例随温度的变化关系

图11 在持续加热过程中不同的温度下完整的Al-CO结构的纳米粒子(原子数量为147)的瞬时形状
对于原子数为55的Al-CO纳米粒子也进行了分子动力学模拟,发现粒子在极低温下(10 K),都不能保持其初始的结构,会转变成ICO结构,说明对于原子数为55的Al-CO结构不是一个能稳定存在的结构.
3 结论
应用分子动力学结合嵌入原子势模型模拟了Fe-ICO、Al-ICO、Al-Dh(原子数量分别为55、147、309、561)和Al-CO(原子数量分别为55、147、309、561)的熔化过程.由于表面原子配位数的减少,表面原子比体原子具有更高的能量,因此在熔化前会有结构的转变.其中原子数量为55、147时Al-Dh、Al-CO结构在熔化前都会发生其结构先转变为ICO结构,原子数为55的Al-CO在极低温下(10 K)都不能维持其初始结构.但是在原子数量为309、561时,升温过程中纳米粒子没有发生结构转变.原子数分别为147、309、561的Fe-ICO的熔化温度分别为1090 K、1190 K、1290 K;原子数分别为147、309、561的Al-ICO的熔化温度分别为570 K、660 K、710 K;原子数分别为147、309、561的Al-Dh的熔熔化温度分别为580 K、670 K、750 K;原子数分别为147、309、561的Al-CO的熔化转变临界温度分别为570 K、660 K、760 K.
