儿童良性癫痫伴中央颞区棘波放电患者的GRIN2A突变筛查及遗传特征分析
2016-11-10刘晓蓉赖锦星江孝建廖卫平黎冰梅
刘晓蓉, 黄 丹, 赖锦星, 余 璐, 孙 辉, 江孝建, 汪 洁, 廖卫平, 黎冰梅
儿童良性癫痫伴中央颞区棘波放电患者的GRIN2A突变筛查及遗传特征分析
刘晓蓉, 黄 丹, 赖锦星, 余 璐, 孙 辉, 江孝建, 汪 洁, 廖卫平, 黎冰梅
目的 对儿童良性癫痫伴中央颞区棘波放电(BECTS)患者进行GRIN2A基因突变筛查,分析其在BECTS患者中的致病性和遗传特征。 方法 收集2012年6月~2014年12月期间我院诊断的BECTS患者,采用PCR扩增和一代测序方法筛查GRIN2A基因突变。结果 在收集的53例BECTS患者中,发现4例存在GRIN2A杂合突变,其中1例患者同时具有2个GRIN2A基因突变位点。突变包括4个错义突变(c.2627T>C,p.I876T;c.1341T>A,p.N447K;c.4322C>A,p.T1441N;c.1271C>T,p.P424L)和1个剪切位点突变(c.415-38C>T),且均为未见报道的新突变位点。除c.1271C>T为新生突变、c.1341T>A未能验证突变来源外,其余3个突变均为遗传性突变。突变率为7.5%,外显率为75%。经保守性分析,发生错义突变的氨基酸变化位点均为高度保守。结论 GRIN2A可能是BECTS的致病基因。GRIN2A突变存在不完全外显,临床进行遗传咨询时应引起高度重视。
伴中央颞区棘波的儿童良性癫痫; GRIN2A; 基因型; 表型
儿童良性癫痫伴中央颞区棘波放电 (benign childhood epilepsy with centrotemporal spikes,BECTS),又称Rolandic癫痫,是一种常见的儿童遗传性局灶性癫痫,占儿童癫痫的15%~24%[1]。
近年来研究表明:在包括BECTS在内的中央颞区棘波放电癫痫谱系患者中,存在离子型N-甲基D-天门冬氨酸受体2A基因(glutamate receptor,ionotropic,N-methyl D-aspartate 2A,GRIN2A)基因突变,而后者很可能是BECTS的致病基因[2~4]。 目前尚未见有关中国BECTS患者GRIN2A基因突变的研究报道。本研究中,我们首次对中国汉族BECTS患者进行GRIN2A基因突变筛查,分析其突变及遗传特点,以探讨GRIN2A在BECTS中致病性。
1 研究对象和方法
1.1 研究对象 收集2012年6月~2014年12月期间在广州医科大学附属第二医院癫痫专科就诊的中国汉族BECTS患者。根据Tsai的描述[5]和ILAE-1989有关癫痫和癫痫综合征分类[6]进行BECTS的诊断,具体标准为:(1)起病年龄为2~14岁。(2)癫痫发作类型为部分性发作(包括单纯部分性和复杂部分性发作),或继发全身强直阵挛发作,发作与睡眠关系密切,多于入睡后不久和清晨将醒时发作。(3)体格检查无明显神经系统局灶性体征。(4)脑电图背景活动正常,单侧或双侧中央-中颞区局限性棘波或尖波放电,睡眠期放电增多。(5)影像学检查未见与癫痫相关的结构性损害。(6)抗癫痫药物治疗有效,预后好。
1.2 GRIN2A基因突变筛查 在获得患者或监护人知情同意后用EDTA抗凝管采集患者静脉血4 ml,采用美国基因技术有限公司提供的QuickGene-610L及QuickGene mini80基因组DNA快速提取试剂盒抽提DNA。根据Ensemble检索(http://asia.ensembl.org)的基因序列设计GRIN2A基因外显子PCR引物设计(见表1)。用PCR方法对GRIN2A基因全部外显子扩增。而后用AppliedBiosystems-3730XL测序分析仪测序,并使用Vector_NTI_v8.0生物学软件进行数据分析,确定有无基因变异。同时以100个健康体检的正常人做对照,以确定所发现的基因变异是否为基因突变。
2 结 果
共收集53例BECTS患者,其中男性35例,女性18例。经过基因筛查,发现4例患者存在5个GRIN2A基因的杂合突变(见图1),突变率为7.5%(4/53)。突变位点包括4个错义突变(c.1271C>T,p.P424L,c.1341T>A,p.N447K;c.2627T>C,p.I876T;c.4322C>A,p.T1441N)和1个剪切突变(c.415-38C>T)。其中c.4322C>A和c.1271C>T 为同一患者中具有的两个突变位点。在4个错义突变中,c.1271C>T(p.P424L)和c.1341T>A(p.N447K)位于GRIN2A蛋白的N末端,而c.2627T>C(p.I876T)和c.4322C>A(p.T1441N)位于GRIN2A的C 末端。经查询人类基因突变数据库(http//www.Hgmd.cfac.uk/ac/index.php),上述突变点均为国内外首次报道新突变。且在100例正常对照中未发现上述变异。经氨基酸的蛋白保守性分析表明:I876、N447、T1441、P424氨基酸区域均为保守(见图2)。

表1 GRIN2A 基因PCR扩增引物(转录本号:NM_000833.4)
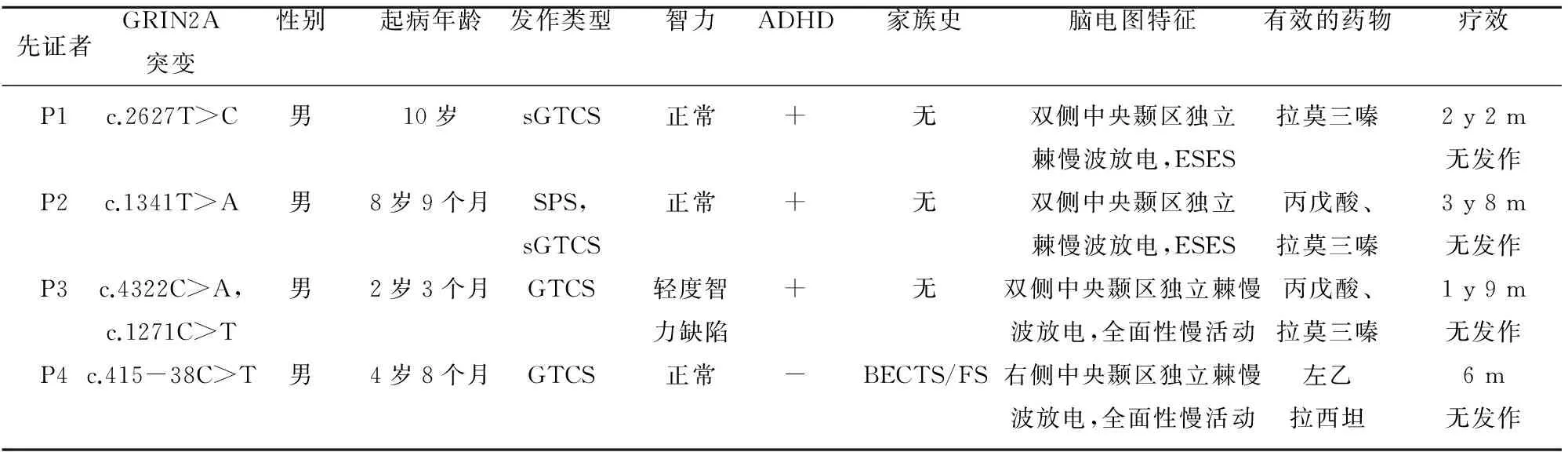
表2 具有GRIN2A基因突变的BECTS患者的临床特征
SPS:simple partial seizure,单纯部分性发作;GTCS:generalized tonic-clonic seizure,全面性强直阵挛发作;sGTCS,:secondary generalized tonic-clonic seizure,继发性全面性强直阵挛发作;BECTS:benign childhood epilepsy with central-temporal spike,良性儿童癫痫伴中央颞区棘波放电;FS:febrile seizure,热性惊厥;ADHD:attention-deficit hyperactivity disorder,注意力缺陷多动症;+:阳性;-:阴性
具有GRIN2A突变患者的临床特征(见表2)。4例患者均为男性。平均起病年龄为6.42岁(2岁3个月~10岁),4例患者均有强直-阵挛发作。除具有双突变的患者(P3)有轻度的智能障碍外,其余患者的智力均正常。在4例患者中,3例患者曾被诊断为注意力缺陷多动障碍。仅1例患者(P4)有热性惊厥和BECTS家族史,其余3例患者在临床上均表现为散发(见图2)。3例患者(P1~P3)表现为双侧中央-中颞局灶性癫痫样放电,仅1例(P4)为单侧放电。2例患者脑电图表现为慢波睡眠期电持续状态。另2例患者除局限性癫痫样放电外还表现为清醒期和睡眠期阵发性弥漫性高幅delta活动。4例患者对抗癫痫药物反应较好,丙戊酸、拉莫三嗪和左乙拉西坦治疗均有效,所有患者均有半年以上无发作。
从突变来源来看,5个突变位点中,除c.1341T>A(p.N447K)因父母未能抽血而未能验证突变来源,以及c.1271C>T (p.P424L)为新生突变外,其余3个突变均为遗传性突变(见图3)。突变外显率为75%。

图1 BECTS患者GRIN2A基因测序图(WT:野生型;MT:突变型),箭头所指处为突变

图2 GRIN2A的I876T、N447K、T1441和P424L突变位点同源性比较

m/+表示杂合突变,+/+表示不携带GRIN2A基因突变。箭头所示为先证者。BECTS:良性儿童癫痫伴中央颞区棘波;FS:热性惊厥
3 讨 论
尽管BECTS是儿童期最常见的遗传性局灶性癫痫,但临床以散发病例多见,故对其致病基因的定位和致病机制的研究较为困难。近年来研究发现部分BECTS中存在GRIN2A基因突变[4]。但目前尚未见中国BECTS癫痫患者中GRIN2A基因突变的报道。本研究通过对中国汉族BECTS患者的GRIN2A基因筛查,首次发现患者中存在GRIN2A基因突变,且突变位点均为文献中未见报道新突变。
GRIN2A基因位于16p13.2,是N-甲基-D-天门冬氨酸受体(N-methyl-D-aspartate receptors,NMDAR)的NR2A亚基的编码基因。NMDAR是中枢神经系统中参与突触功能调节的神经递质门控离子通道[7],该受体的功能对神经元的兴奋性有重要影响。而NR2A亚基是NMDAR的调节亚基,广泛分布与大脑皮质及海马[8],故可能与癫痫发作有密切关系。目前研究表明GRIN2A基因突变导致的癫痫综合征包括:BECTS、不典型良性部分性癫痫、癫痫伴慢波睡眠期持续放电、Landau-Kleffner综合征、Lennox-Gastaut综合征[9]、早发型癫痫性脑病[10]和不能分类的癫痫伴神经发育异常[4,9,11]。但GRIN2A在不同表型中突变率不同,表型严重的患者中的突变率高,表型轻的患者中突变率低,如在严重的Landau-Kleffner 综合征/ 癫痫伴慢波睡眠期持续放电中突变率为17.6%,在症状相对轻的BECTS患者中突变率只有4.9%[4,11]。在本研究中,BECTS患者的突变率为7.5%,与文献中报道基本一致。本研究中所发现的4个错义突变经过保守性分析均为高度保守,且正常对照中未发现这些基因变异,说明其具有致病性。而且从发育学上来看,GRIN2A在儿童期达到表达高峰,其表达时间与BECTS的起病时间相吻合,说明GRIN2A很可能是BECTS的致病基因。
从突变形式看,本研究中,除1例为剪切位点突变外,其余4例突变均为错义突变,这与文献中报道的一致(错义突变为主,占86.9%)[4]。正常的NMDARs在静息状态下,需甘氨酸与必需亚基NR1、谷氨酸与调节NR2同时结合才能解除Mg2+的阻断、导致Ca2+内流而激活NMDAR。既往功能学研究发现:GRIN2A错义突变可导致Mg2+阻滞作用下降、Ca2+的通透性增加,NMDAR通道开放的平均时间延长,从而使NMDAR功能增强[4]。本研究中所发现的4个错义突变,分别位于NR2A的N末端和C末端,可能通过干扰谷氨酸与NR2A的结合、Mg2+的阻断作用减弱等机制,导致NMDAR受体功能增强,从而使神经元兴奋性增加,导致癫痫的发生。不同突变对NMDAR功能的影响机制和程度还需进一步经功能学研究来证实。
既往研究中发现GRIN2A突变以遗传性突变为主,新生突变仅占19.7%[2~4,11]。本研究中,有类似遗传规律,除c.1271C>T为新生突变外,其余3个可验证来源的突变均为遗传性突变。而GRIN2A的外显率为75%,与文献中GRIN2A的80%的外显率相近[2]。在我们既往有关SCN1A[5]和PRRT2[12]基因的研究中发现,突变的不完全外显性可能与其相对较低的致病性有关。因此,GRIN2A错义突变的致病性很可能相对较低,这也是GRIN2A突变在人群中等位基因频率相对较高的原因。另外,GRIN2A突变的不完全外显现象也是临床上BECTS散发病例较多的原因,对于这一现象的认识对正确地指导临床遗传咨询有重要意义。值得注意的是:本研究中发现了1例复合突变的患者,患者同时具有c.4322C>A 和c.1271C>T两个突变,因此不除外隐性遗传的可能性。这一现象与我们以往有关PRRT2的研究中发现的复合突变相似[12]。突变c.4322C>A来自母亲,但其母亲并未表现出症状,而c.1271C>T为新生突变,很可能后者致病性相对较强。与其他具有单一突变的患者不同,该患者具有轻度智力障碍,故两个突变对NMDAR的影响可能具有叠加作用。
本研究中,3例具有GRIN2A突变的患者有注意力缺陷多动障碍,说明突变不但可引起癫痫,而且对其他高级神经系统活动,如注意力、智力等都有影响。3例具有GRIN2A基因突变的患者表现为双侧中央-颞区癫痫样放电,且2例患者的EEG除局灶性放电外,还表现为阵发性弥漫性慢活动,这与NR2A在双侧大脑半球皮质以及海马分布较多有关。而从胚胎发育学的角度来看,尚无证据表明双侧大脑半球GRIN2A的含量和分布具有差异性,而本研究中1例患者仅表现为单侧中央-中颞放电,其机制不明。对于该患者来说,除进行遗传学方面的病因分析外,还应注意有无局灶性结构性异常的可能性,如局限性皮质发育不良等。
综上所述,GRIN2A可能是BECTS常见的致病基因,其不但可引起癫痫,而且影响患者的认知功能和精神行为。临床对BECTS的患者应进行GRIN2A基因突变筛查,以明确其病因。GRIN2A突变存在不完全外显现象,在进行遗传咨询时应引起高度重视。
致谢:感谢参与患者和患者家属对本研究的无私奉献和支持。
[1]Kramer U,Zelnik N,Lerman-Sagie T,et al. Benign childhood epilepsy with centrotemporal spikes:clinical characteristics and identification of patients at risk for multiple seizures[J]. J Child Neuro,2002,17:17-19.
[2]Lesca G,Rudolf G,Bruneau N,et al. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction[J]. Nature Genetics,2013,45:1061-1066.
[3]Carvill GL,Regan BM,Yendle SC,et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders[J]. Nature Genetics,2013,45:1073-1076.
[4]Lemke JR,Lal D,Reinthaler EM,et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes[J]. Nature Genetics,2013,45:1067-1072.
[5]Tsai MH,Vears DF,Turner SJ,et al. Clinical genetic study of the epilepsy-aphasia spectrum[J]. Epilepsia,2013,54:280-287.
[6]Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy[J]. Epilepsia,1989,30:389-399.
[7]Cull-Candy S,Brickley S,Farrant M. NMDA receptor subunits:diversity,development and disease[J]. Cur Opin in Neurobiol,2001,11:327-335.
[8]Laurie DJ,Bartke I,Schoepfer R,et al. Regional,developmental and interspecies expression of the four NMDAR2 subunits,examined using monoclonal antibodies[J]. Brain Res Mole Brain Res,1997,51:23-32.
[9]Reutlinger C,Helbig I,Gawelczyk B,et al. Deletions in 16p13 including GRIN2A in patients with intellectual disability,various dysmorphic features,and seizure disorders of the rolandic region[J]. Epilepsia,2010,51:1870-1873.
[10]Yuan H,Hansen KB,Zhang J,et al. Functional analysis of a de novo GRIN2A missense mutation associated with early-onset epileptic encephalopathy[J]. Nature Communications,2014,5:3251.
[11]Endele S,Rosenberger G,Geider K,et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes[J]. Nature Genetics,2010,42:1021-1026.
[12]Liu XR,Wu M,He N,et al. Novel PRRT2 mutations in paroxysmal dyskinesia patients with variant inheritance and phenotypes[J]. Genes Brain & Behavior,2013,12:234-240.
GRIN2A Mutations and Genetic Features in Benign Childhood Epilepsy with Centrotemporal Spikes
LIUXiaorong,HUANGDan,LAIJinxing,etal.
(InstituteofNeuroscienceandtheSecondAffiliatedHospitalofGuangzhouMedicalUniversityandKeyLaboratoryofNeurogeneticsandChannelopathiesofGuangdongProvinceandtheMinistryofEducationofChina,Guangzhou510260,China)
Objective To screen GRIN2A mutations in Chinese Han patients with benign childhood epilepsy with centrotemporal spikes (BECTS) and analyze the features of GRIN2A mutations. Methods Patients with BECTS were recruited. GRIN2A mutations were screened by PCR and direct sequencing. Results Fifty-three patients with BECTS were recruited. Five new heterozygous mutations in GRIN2A were identified in 4 patients with BECTS,including 4 missense mutations (c.2627T>C,p.I876T;c.1341T>A,p.N447K;c.4322C>A,p.T1441N;c.1271C>T,p.P424L) and 1 splicing site mutation (c.415-38C>T). Among the patients with GRIN2A mutations,one of the patients had two mutations (c.4322C>A and c.1271C>T). Except for c.1271C>T was a de novo mutation and c.1341T>A was not available for inherited analysis,other mutations were inherited. The rate of incomplete penetrance was 75%. Conclusion GRIN2A is a causative gene for BECTS. Screening of GRIN2A mutation is highly recommended in the patients with BECTS. Incomplete penetrance should be paid attention in the area of genetic counselling.
Benign childhood epilepsy with centrotemporal spikes; GRIN2A; Genotype; Phenotype
1003-2754(2016)01-0022-05
2015-11-10;
2015-12-22
广东省教育厅高等学校科技创新项目(No.2012KJCX009);广州市科信局科技计划项目(No.2014J4100062、No.201508020011);国家自然科学基金项目(No.81571273)
[广州医科大学附属第二医院神经内科,广州医科大学神经科学研究所(广东省重点实验室,神经遗传与离子通道病省部共建教育部重点实验室)广东 广州 510260]
刘晓蓉,E-mail:happyxiaorongo@163.com
R742.1
A
