基于有限突变模型和大规模数据的19个常染色体STR的实际突变率研究
2021-11-01刘志勇任贺陈冲张京晶张晓梦石妍石林玉陈滢程凤贾莉陈曼范庆炜张家榕李万婷王萌春任子林刘雅诚倪铭孙宏钰严江伟
刘志勇,任贺,陈冲,张京晶,张晓梦,石妍,石林玉,陈滢,程凤,贾莉,陈曼,范庆炜,张家榕,李万婷,王萌春,任子林,刘雅诚,倪铭,孙宏钰,严江伟
研究报告
基于有限突变模型和大规模数据的19个常染色体STR的实际突变率研究
刘志勇2,任贺3,陈冲4,张京晶5,张晓梦1,石妍4,石林玉1,陈滢4,程凤1,贾莉4,陈曼6,范庆炜7,张家榕1,李万婷1,王萌春1,任子林8,刘雅诚4,倪铭8,孙宏钰2,严江伟1
1.山西医科大学法医学院,太原 030001 2.中山大学中山医学院法医学系,广州 510080 3.北京警察学院,北京 102202 4.北京通达首诚司法鉴定所,北京 100192 5.北京华彦科技有限公司司法鉴定所,北京 100192 6.南方医科大学法医学院,广州 510515 7.川北医学院法医学系,南充 637000 8.北京辐射医学研究所,北京 100850
短串联重复序列(short tandem repeat, STR)已广泛用于法医学亲子鉴定和个体识别中,但STR的突变可能会影响其结果的解释。在大多数类似研究中,由于忽略“隐性”突变现象,STR的突变率被低估。鉴于此,为获得更加准确的STR实际突变率,本研究使用Slooten与Ricciardi提出的有限突变模型和大规模数据,对28,313例(78,739个体)中国北京汉族已确认亲生关系的亲子鉴定案的20个常染色体STR基因座(和;由于有限突变模型中未包含的矫正参数,因此本文实际计算其余19个STR基因座的突变率)进行了调查。结果发现,所有基因座均存在突变现象,总计发生1665个突变事件,包括1614个一步突变,34个两步突变,8个三步突变和9个非整步突变。基因座特异性的平均实际突变率在三联体中为0.00007700 ()~0.00459050 (),在二联体中为0.00000000 ()~0.00344850()。此外,本研究还分析了表面和实际突变率、三联体和二联体突变率、父源和母源的突变率之间的关系。研究表明,实际突变率多大于表面突变率,而且1*/2* (表面突变率)的比值通常也大于1/2 (实际突变率) (1*,1;2*,2分别是一步和两步的突变率),即更多的“隐性”突变被释放出来。而且父源和母源的三联体和二联体的突变率也有存在差异。随后,将这些突变率数据与已发表的中国其他汉族人口的相关研究进行比较,展现出了STR突变率的时间与区域差异。由于样本量大,本研究中还报告了一些少见的突变事件,例如同卵双胞胎突变和“假四步突变”等。综上所述,本研究通过大量数据获得了接近真实的STR突变率的估计值,不仅可为中国法医DNA数据库和群体遗传学数据库提供重要的基础数据,也对开展法医学个体识别、亲权鉴定和遗传学研究具有重要的意义。
常染色体STR;有限突变模型;亲子鉴定;中国汉族人群;突变分析
STR (short tandem repeat)是人类基因组中非常丰富的一类遗传标记[1],由于其高度的多态性[2,3],被广泛应用于个体识别、亲子鉴定和群体遗传学等领域。然而,在大多数STR基因座中都可以观察到突变事件,一般认为复制滑脱是导致STR突变的主要机制[4]。这些突变现象往往会对法医学实践中证据权重评价产生影响,比如在计算累积父权指数(cumulative paternity index, CPI)时需要特别考虑突变现象,过高过低的突变率数据都不利于得出客观结论,因此获得STR基因座的准确突变率是非常重要的。
STR基因座突变率的实际评估计算中,往往会受抽样群体大小的影响[5]。尽管已有研究者发表大量关于STR基因座突变率的论文,但基于较少的样本量,其STR突变率的计算结果可能存在偏差。除了以上因素外,突变率的不同计算方法也会对STR实际突变率计算产生影响。在已报道的多数研究中STR突变率一般是通过直接计数法来计算的5~8],并且将三联体和二联体的数据进行了合并。然而,该方法没有考虑“隐性突变(hidden mutation) ”现象,因为人们普遍认为当发生突变时,突变步数越少越真实,但实际并非总是如此,即某些突变会被隐藏。鉴于通过三联体和二联体的计算得到的STR基因座突变率有一定差异,因此合并三联体和二联体的突变数据可能是不合理的。即,直接计数法对于STR的实际突变率的估计有一定偏差。
针对以上抽样群体尺度与计算方法的问题,本研究采用了大量亲子鉴定数据和Slooten与Ricciardi[9]的更为严谨的有限突变模型计算方法。基于28,313例已确认亲生关系的中国汉族人群亲子鉴定案例,分别计算了三联体和二联体两种情形的19个常染色体STR基因座实际突变率。本文详细比较了表面突变率(直接计数法)和实际突变率(有限突变模型),并与其他多个群体的STR突变率进行了对比分析。最后,本文还讨论了影响STR突变率的几个因素,比如突变年龄与来源、等位基因长度、基因座杂合度等。
1 材料与方法
1.1 数据来源
本研究涉及的基因座突变数据来自2009~2019年间中国北京汉族人群的常规亲子鉴定案例的统计结果,该数据已获得北京通达首诚司法鉴定所许可使用。所有数据均采用匿名化处理,不涉及个体相关除突变信息以外的其他STR基因座具体分型,也不涉及家系情况。本次统计分析的亲子鉴定案例的CPI值均大于10,000,达到亲子鉴定的确认标准(未能确认的案例被除外),且突变的STR基因座已经过多个试剂盒验证。总计28,313例(78,739个体)亲子鉴定数据被纳入,包括22,113例三联体(trio’s)和6200例二联体(duo’s) (父亲–孩子:2342例;母亲–孩子:3858例)。该数据涉及的STR基因座包括CODIS 系统()和。由于有限突变模型[9]中未包含基因座的计算矫正参数,因此本研究中共包括除之外其余19个STR基因座的突变率计算。
1.2 突变率计算
在已确认亲生关系的亲子鉴定案例中,当发现STR等位基因违反孟德尔遗传定律时,认为该STR基因座发生突变事件[10]。在计算过程中,突变步数与来源的判定根据Brinkmann等[10]和Weber等[11]的描述,本研究中所有可疑的STR突变事件均再次通过同一试剂盒和其他试剂盒进行检测,确保分型正确。所有STR基因座的三联体和二联体父源母源突变事件的统计结果见附表1。在突变率计算过程中,一方面使用直接计数法公式*=n/N计算表面突变率,其中n是观察到的突变事件的数目,N为减数分裂的发生次数;另一方面在得到表面突变率后,通过概率矫正因子矩阵A矫正得到实际突变率(见方程(I))。其中A_{k, l}代表观测到k步突变,实际是l步突变的概率。对于任一A_{k, l},Slooten和Ricciardi[9]基于NFI的参考数据集(2085个体)的等位基因型频率,随机组合一对夫妇,模拟生成子代的基因型,其中从母亲遗传一个没有突变的等位基因,但从父亲遗传得到一个实际有l步突变的等位基因,即已知实际突变步数,然后通过观察得到表面突变步数。上述过程通过计算机模拟重复100,000次,计算不同突变步数的频率,进而估计最终突变转移概率A_{k, l}。该模型假设:(1)当发生不确定来源的突变时,认为父源与母源发生的概率相同,均为0.5;(2)当观察到的突变事件可以同时解释为相同的突变增加或者突变减少时,该模型方法不予区分。本文采用模型中下列矩阵方程(I)计算实际突变率。

在“方程(I) ”中,k*为表面为k步的突变率,0*为表面不发生突变的概率;k代表实际为k步的突变率,0为实际不发生突变的概率;Ak,l是表面突变步数为k,而实际步数为l的概率;*为基因座特异性表面突变率;为基因座特异性实际突变率。为了便于理解该模型,方程(Ⅱ)列出了方程(I)的展开式。以TH01为例,统计计算得到了表面突变率k*后,再根据Slooten和Ricciardi[9]提供的矫正矩阵A_{k, l},可以结合方程(Ⅱ)反解出实际突变率k,其具体的计算过程见附表2。本文采用在线计算器(https://www.numberempire.com/matrixbinarycalculator. php)进行矩阵求逆。
方程(Ⅱ)
2 结果与分析
2.1 突变分析
本项研究中,在28,313例确认的亲子鉴定案例(22,113例三联体和6200例二联体)的20个常染色体STR基因座中共发现了1665个突变事件,所有基因座均观察到突变现象。基于有限突变模型计算得到的三联体和二联体的基因座特异性实际突变率和总突变率数据显示(表1,不包含基因座):三联体基因座特异性平均实际突变率范围为0.00007700 ()~0.00459050 (),二联体为0.00000000 ()~0.00344850 ();基因座具有较高的父源突变率(0.00767100)和总突变率(0.00459050),具有较高的母源突变率(0.00237900),与Jin等[12]基于直接计数法的研究结果一致。
同卵双胞胎(monozygotic, MZ)是由单个精子和单个卵子结合形成受精卵后,分裂成两个胚胎形成的,其发生率约为1/250[13]。从理论上讲,同卵双胞胎拥有相同的基因组,然而研究者在STR[14]、拷贝数变异(CNV)[15]和单核苷酸多态性(SNP)[16]中观察到了一些差异。本次研究中,在10对MZ中发现了相同突变,包括7例男性和3例女性(附表3),突变基因座为(2例)、(2例)、(2例)、、、和,且均为一步突变。在已公开发表的论文中,尚未报道同时在MZ中发现不一致的STR突变,但Wang等[17]报道了一对MZ中其中一个个体的基因座出现三等位基因模式的案例。在亲子鉴定实践中,一般较少会在同一个体观察到多个突变。如果观察到,其可能会干扰鉴定人得出亲子关系的正确结论。在本次研究的28,313例亲子鉴定案件中,共发现27例孩子的基因型中可观察到2个突变的事件,发生率为0.00095363 (27/28,313,20个常染色体STR基因座) (附表4A);同时也发现3例孩子的基因型中可观察到3个突变的事件(附表4B)。在这30例的多基因座突变案例中,本研究使用了总计超过39个基因座的多个STR试剂盒对亲子关系进行了确认。
2.2 表面与实际突变率的比较
基于直接计数法和有限突变模型,计算得到每个STR基因座的表面突变率(*)与实际突变率(),三联体的相关突变率数据见附表5,二联体相关数据见附表6。在使用有限突变模型的矩阵计算(方程(I))中,本研究发现有三个负值的实际突变率,即三联体组的和基因座的M2;二联体组的基因座的F1(M,F分别代表母源和父源突变)。为了查找原因,本研究分析了和基因座在三联体中的突变特征,发现母源突变中可观察到一步和三步突变,但未观察到两步突变;同理,二联体组的基因座仅在父源中观察到两步突变,但未观察到一步突变。即表明,实际突变率负值可能是在某STR基因座可观察到高步数突变,但未观察到某个低步数突变的情况下发生的(即非连续的突变步数现象)。换言之,即这几个负值可能在提示潜在低步数突变应该会被观察到,但实际中并没有观察到,这已经超过了有限突变模型的可解释范畴。因此在数据处理过程中,把三个负值实际突变率设为零。如图1所示,在父源和母源突变之间有一个明显的界限,特别是在三联体的一侧(图1A),这可能提示三联体与二联体的计算得到的STR突变率是有差异的,尽管这种差异可能来自于三联体与二联体本身,或者来自于样本量的差异,但可间接说明以往直接合并三联体与二联体数据进行计算存在不合理之处。此外,从该图也可发现在三联体和二联体中,和基因座的表面和实际突变率都较低。与之相反的是,和基因座在三联体中的表面和实际突变率均较高,而和基因座在二联体中的表面和实际突变率较高。为了进一步分析有限突变模型的校正效果,绘制了实际突变率与表面突变率比值图(/*) (图2)。该图表明,除父源的和(/*≈1)和母源的基因座(/*≈1)外,其余基因座的突变率均得到了较大程度的矫正(/*>1)。就矫正程度而言,二联体组的基因座的效应表现得更为明显(与Slooten和Ricciardi[9]的研究结果相似),尤其是和基因座。也就是说有限突变模型通过参数矫正的方式,将更多的隐性的突变挖掘出来。然而,在亲子鉴定过程中,需要计算有突变率参与的父权指数时,目前一般采用直接计数法计算得到平均STR突变率(比如男性取0.002;女性取0.001~0.0005)来进行。若为了更严谨的计算,可采用三联体和二联体的父源、母源或者STR基因座特异性突变率,当然也可以使用本次研究基于有限突变模型计算获得的STR基因座特异性实际突变率。从理论上讲,计算单个基因座父权指数时,采用上述两种算法得到的突变率,结果是会存在差异,但对总体的鉴定结论的影响有限。

表1 三联体与二联体STR基因座特异性实际突变率
“”表示实际突变率;“F”表示父源;“M”表示母源。

图1 三联体和二联体表面和实际突变率热图
A:三联体情形;B:二联体情形;“*”表示表面突变率;“”表示实际突变率。

图2 表面突变率与实际突变率比较
“A”和“B”代表三联体组父源(F)和母源(M)的/*比值;“C”和“D”分别代表二联体组父源(F)和母源(M)的/*比值;空白处表示比式的分母为零的情况。
2.3 三联体和二联体组突变率的比较
在以往基于直接计数法研究中,一般将三联体和二联体组的数据进行合并计算突变率。然而,三联体和二联体得到的突变率水平是有一定差异,其在2.1和2.2中已进行部分阐述。在大多数STR基因座中,三联体父源的实际突变率值大于二联体的父源相关值(图3,绿色>黑色);三联体母源的实际突变率值大于二联体的母源相关值(图3,粉色>蓝色);它们的比值表现出相似的规律(图3),特别是在基因座。与此同时,本研究也发现存在相反趋势的基因座,如三联体组的、和二联体组的等,因此将三联体和二联体的数据合并是不合理的,它们之间的差异是存在且不宜忽略的。
2.4 父源和母源突变率的比较
在1665个突变事件中,共观察到1178例父源突变和293例母源突变事件,同时有194例突变来源不明(父源或母源),其中父源突变占多数,为70.75% (图4A)。在三联体组中,总F为0.06405550,M为0.01937639 (表1);二联体中,总F为0.05146300,M为0.01220500 (表1)。从以上数据可以得知,父源突变率一般较母源突变率高,在三联体和二联体组中分别约为3.3倍和4.2倍,其与以往的研究结果一致[11,12]。Jónsson等[18]对冰岛1548个三联体进行的新生突变(de novo mutations, DNMs)研究也表明父源的突变数量约是母源的4倍,这也侧面反映了本次研究的结果。本研究还比较了不同突变来源的基因座特异性突变率(图5)。如图5A显示,三联体中绝大多数基因座不同突变来源计算得到的突变率具有一定差异性,除和基因座外,父源突变率是母源突变率2倍以上。而二联体组也显示了相似的趋势(图5B)。这可能是男性与女性形成配子时经历的细胞分裂次数不同,而男性需要产生更多的配子的缘故[10]。可以预知,使用突变来源特异性基因座突变率将有助于亲子关系等法医学鉴定得出的更加客观而准确结论。另外,为了说明不同突变来源的年龄范围,本研究制作了父源(图4B)和母源(图4C)突变年龄的扇形图,从中可以看到父源突变的年龄范围比母源突变的年龄范围更广,这可能是由于男性在较长时间内可以保持一定的生殖能力,而女性不可以的原因。

图3 三联体和二联体中Fμ、Mμ及其比值的比较
“trio’s”表示三联体组;“duo’s”表示二联体组;空白处表示比式的分母为零的情况。
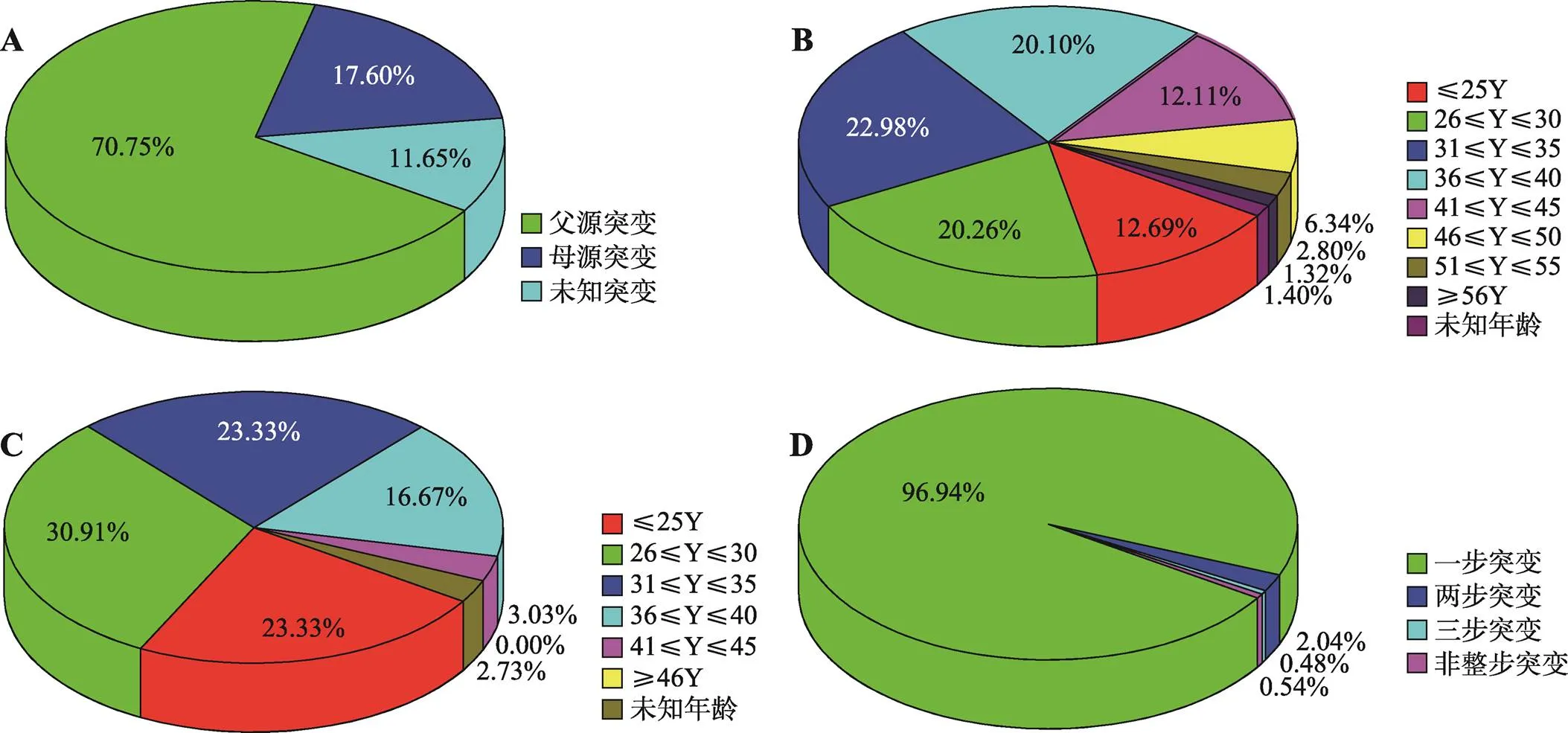
图4 本次研究中突变事件来源、年龄和步数的比较
A:突变来源分布;B:父源突变年龄分布;C:母源突变年龄分布;D:突变步数分布。
2.5 突变步长与突变方向分析
在所有的突变事件中,共观察到1614个一步突变、34个两步突变、8个三步突变和9个非整步突变。一步突变占总突变事件的96.94%,是主要的突变类型,逐步突变模型(stepwise mutation model, SMM)可以解释一步突变为主的现象[19,20]。在其他非一步突变中,两步突变占2.04%,三步突变占0.48%,而非整步突变占0.54% (图4D)。发生非整步突变的基因座分别为:(3例)、(2例)、、、和;突变来源为父亲的6例,母亲的1例,未能确定突变来源的2例;非整步的突变方向,其中7例突变减少,1例突变增加,另有1例不能确定突变方向;关于非整步突变的步数,0.1、0.2、0.3步均有观察到,其中有5例含有0.2步突变,2例有0.3步突变,1例有0.1步突变,另有1例为0.3或者0.1步突变。就本研究观察到的9例非整步突变而言,突变减少和包含0.2步突变是占多数的,详细信息展现在附表7中。为了进一步分析有限突变模型对于STR基因座等位基因不同突变步数的突变率影响,本研究计算了三联体中发生一步突变与两步突变的比值(图6)。在父源(图6A)中,1*/2*的值普遍大于1/2,特别是在基因座(本次研究:1*/2*≈104;1/2≈73),这表明由于有限突变计算模型的使用,1/2之间的差异正在减小。这些规律与Slooten与Ricciardi[9]的研究相似(1*/2*≈75;1/2≈58),也就是说更多的高步数隐性突变被挖掘出来。在母源(图6B)基本上也显示了这一规律,但发现了具有相反趋势的基因座,如、和,根据该模型作者的解释[9],这可能是由于模型本身不完美的原因造成的。
尽管有限突变计算模型中不考虑突变方向(突变减少/增加),但本研究仍然统计了等位基因减少和增加的分布情况。在1665次突变事件中(附表8),总计有706次突变减少(mutation loss),703次突变增加(mutation gain),256次为不确定突变方向或者分组;突变减少与增加(loss/gain)之比约为1∶1,与Lan等[21]的研究相似。Brinkmann等[10]报道认为突变的发生与等位基因的长度存在一定关联,长的等位基因比短的等位基因更易于发生突变;Shao等[5]认为较长的等位基因倾向于发生突变减少事件,而短的等位基因则更倾向于发生突变增加事件。为了探究等位基因长度与突变方向之间的关系,参照Ge等[22]和Xu等[23]的方法,将每个STR基因座的等位基因根据长度分为三组,即较短(short group)、中等(medium group)、较长(long group)等位基因组,详细的统计数据见附表8。从表中可计算得知在以上三个分组中,总的突变次数分别为105、1042、397,其中中等等位基因长度组发生的突变事件最多(约为67.49%),较长等位基因长度组次之(约为25.71%),而较短等位基因长度组最少(约为6.80%),也即中等及较长等位基因更易于发生突变,较短的等位基因发生突变较少,这与Xu等[23]的研究结论一致;较短等位基因组突变减少与增加(loss/gain)的比值约为0.49 (<1),中等组为0.86,较长组为1.72 (>1),本研究结果也表现出等位基因“短变长,长变短”的规律,这也得到Shao等[5]、Ge等[22]、Xu等[23]、Dupuy等[24]的支持,其可能与STR的序列结构有关。

图5 不同突变来源的基因座特异性突变率比较
A:三联体情形;B:二联体情形;空白处表示比式的分母为零的情况。

图6 三联体父源与母源的突变步长分析
A:父源情形;B:母源情形;空白处表示比式的分母为零的情况。
2.6 杂合度与实际突变率的关系
一般认为,杂合度高的基因座其突变率也较高,也即突变率高可能是基因座杂合度高的一个原因。据此根据已有文献报道的杂合度数据[25],分析了每个STR基因座的杂合度和实际突变率的关系,使用Origin软件绘制线性拟合图(图7),Pearsonʼs= 0.68454。结果也相似地表明,杂合度较低的STR基因座突变率普遍较低,如、基因座,这与前人的研究相一致[12]。
3 讨论
不同的突变率计算方法结果可能存在潜在的偏差,比如传统的直接计数法在处理突变数据时未考虑到“隐性突变”现象[26,27]。此处以两个确认的三联体亲子鉴定为例进行说明(下文“a”为正整数)。案例一:某STR基因座母亲的分型是M=(a, a+1),孩子是C=(a, a+1),父亲是F=(a–1, a+1),假设真实的遗传情况为孩子C的“a+1”来自母亲M的“a+1”,“a”来自父亲F的“a–1”或者“a+1”一步突变后的“a”,即实际发生一步突变。从表面上看,可能会认为孩子C的“a+1”遗传自父亲F的“a+1”,而“a”遗传自母亲M的“a”,此时表面上会认为没有突变发生,即“突变发生有无认识偏差”型隐性突变(Type Ⅰ型);案例二:某STR基因座母亲的分型是M=(a+1, a+1),孩子是C=(a, a–1),父亲是F=(a, a–1),假设真实遗传情况为孩子C的“a–1”来自母亲M的“a+1”两步突变后的“a–1”,“a”来自父亲F的“a”,即实际发生两步突变。从表面上看,可能会认为孩子C的“a–1”遗传自父亲F的“a–1”,“a”遗传自母亲M的“a+1”一步突变后的“a”,此时表面上会认为发生一步突变,因为当不知道突变真相时,一般判断为较短的突变步数,即“真实突变步数认识偏差”型隐性突变(Type Ⅱ型)。类似地,隐性突变也可以出现在二联体中。在上述案例中,表面观察到的突变被定义为表面突变(apparent mutation, ApM),而不可见的真实突变被定义为实际突变(actual mutation, AcM)。因此,如果不考虑隐性突变(比如直接计数法),突变率可能就会被低估。尽管研究者无法直接从表面的STR分型突变现象中洞察真实的突变,但Slooten和Ricciardi[9]通过突变模拟的方式研究了该隐性突变现象,并提供了可接近真实突变率的计算模型和矫正参数。该模型根据突变的等位基因是否超过STR等位基因分型标准品(ladder)的范围L,可分为有限突变模型(restricted mutation model)和无限突变模型(unrestricted mutation model),而真实的生物学过程可能介于这两个模型之间。当不知道突变的等位基因是否超过Ladder时,无限突变模型可能是合适的。然而,基于大量数据研究调查发现[9],突变的等位基因不包括在Ladder中是一种非常少见的现象,本次研究的数据也证明这一点。综合上述,鉴于使用简单的有限突变模型是具有一定合理性的,本研究拟采用该模型计算实际STR突变率。
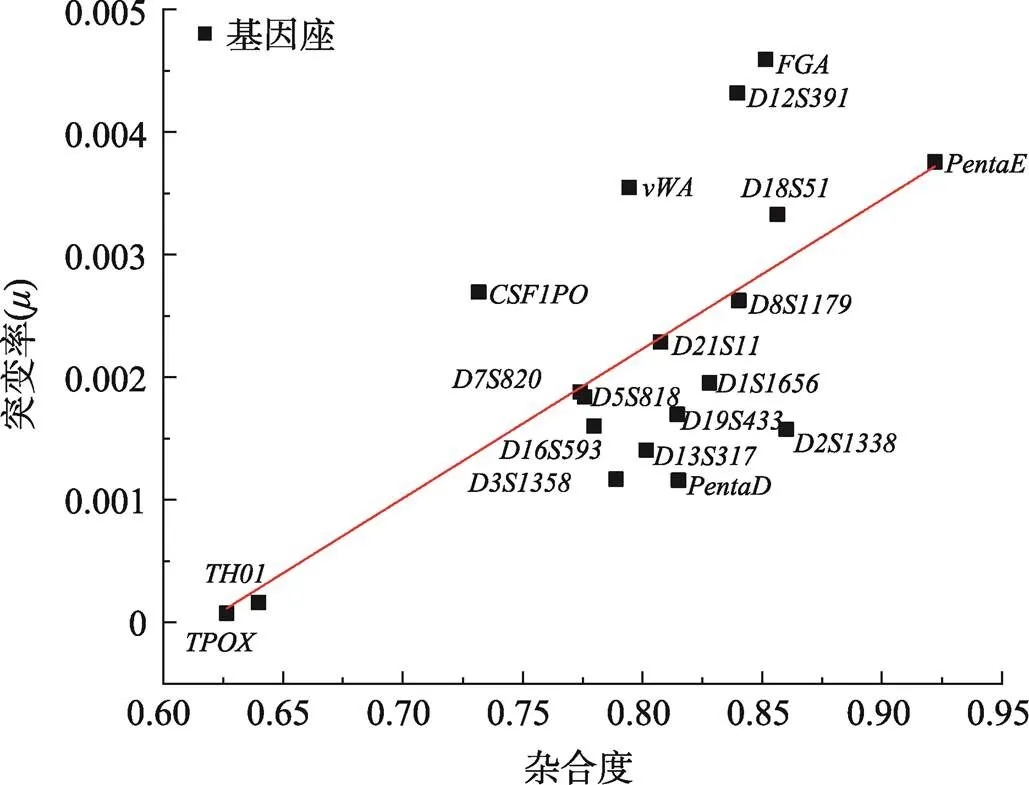
图7 19个常染色体STR基因座杂合度与实际突变率的关系
基于以上有限突变模型计算获得的STR基因座实际突变率,本研究一方面为了观察其是否有时间差异,收集了Yan等[28]于2006年报道的与本研究具有相同来源的北京汉族突变率数据,这组数据调查了6532例亲子鉴定案例中的19,754个样本的17个常用STR基因座特异性突变率, 这些基因座均包含在本次研究中;另一方面为了观察本研究的北京汉族与中国其他汉族人群突变率的地域差异,收集了中国其他地区或省(华北、华东、华南地区和河南、上海、贵州、河北、云南、福建、广东等省)汉族群体的突变率数据(附表9)。如各基因座突变率比较的散点图所示(图8),在时间变化方面:共享的17个STR基因座中,除和外,其余13个基因座的突变率在14年间中增幅较大(红色方框与黑色圆点),尤其是和的突变率增加将近一倍;在地域变化方面,不同汉族群体间的STR基因座特异性突变率具有一定差异,特别是在和基因座,这些差异可能来自于多个地域来源样本的遗传结构不同[29,30],或是使用群体样本量不同,或使用的计算模型方法不同。
亲子鉴定实践中,有时可能遇到假突变现象(fake mutation),其应引起足够的重视。例如,本次研究中在一例确认的三联体亲子鉴定中基因座发现其基因型不符合遗传规律的现象,称之为“假四步突变(fake four-steps mutation) ”:假设父AF=8,母亲M=12和孩子C=8 (图9)。当使用四个STR扩增试剂盒(PowerPlex®21 (Promega,美国)、MicroreaderTM21 (阅微基因,北京)、MicroreaderTM23sp-B (阅微基因,北京)、AGCU 17+1 system (中德美联,无锡))确认基因型后,得到相同的结果。起初,怀疑母亲和孩子之间可能发生了四步突变,即母亲的等位基因“12”突变为“8”。为了进一步证实这个猜想,首先使用ForenSeq DNA Signature Prep Kit (Verogen, 美国)在MiSeq FGx仪器(Illumina,美国)中进行测序,并使用ForenSeq通用分析软件(Verogen,美国)分析原始数据。测序结果显示(图9),母亲的等位基因“8”发生了丢失,其序列与孩子的等位基因“8”是一致的。为了继续寻找母亲等位基因丢失的原因,使用primer5软件设计了基因座的引物(F:TGGAAGCTGCATAGAC;R:ACTCCATACAAATGCAAGAGA),并进行了克隆测序(图9)。如红色方框所示(图9),母亲的等位基因“8”的核心区上游第17 bp处发生单碱基突变,由正常的“C”突变为“T”。以上研究表明,基因座引物结合区的突变可能是多个试剂盒未能成功扩增出母亲等位基因“8”的原因。实际中,真正的四步突变是非常少见的,当遇到可疑的四步突变时,测序有时是很必要的。

图8 中国10个不同地区或省汉族人群突变率比较

图9 基于三种方法对D13S317基因座的测序结果
本研究在28,313例确认的亲子鉴定案例中,共发现1665个突变事件。基于以上突变数据和有限突变模型,分别计算了三联体和二联体案例中的父源与母源的19个常染色体STR基因座的特异性实际突变率。与直接计数法计算突变率相比,本次的计算结果揭示了更多的隐性突变现象,因此获得的突变率数值理论上更加接近真实情况。本研究不仅可为中国法医DNA数据库和群体遗传学数据库提供重要的基础数据,也对开展法医学个体识别、亲权鉴定和遗传学研究具有重要的意义。
附加材料详见文章电子版www.chinagene.cn。
[1] Subramanian S, Mishra RK, Singh L. Genome-wide analysis of microsatellite repeats in humans: their abundance and density in specific genomic regions., 2003, 4(2): R13.
[2] Bär W, Brinkmann B, Budowle B, Carracedo A, Gill P, Lincoln P, Mayr W, Olaisen B. DNA recommendations. Further report of the DNA Commission of the ISFH regarding the use of short tandem repeat systems. International Society for Forensic Haemogenetics,, 1997, 110(4): 175–176.
[3] Butler JM. Short tandem repeat typing technologies used in human identity testing., 2007, 43(4): 2–5.
[4] Kornberg A, Bertsch LL, Jackson JF, Khorana HG. Enzymatic synthesis of deoxyribonucleic acid, XVI. oligonucleotides as templates and the mechanism of their replication., 1964, 51(2): 315–323.
[5] Shao CC, Lin MX, Zhou ZH, Zhou YQ, Shen YW, Xue AM, Zhou HG, Tang QQ, Xie JH. Mutation analysis of 19 autosomal short tandem repeats in Chinese Han population from Shanghai., 2016, 130(6): 1439–1444.
[6] Li HX, Peng D, Wang Y, Wu RG, Zhang YM, Li R, Sun HY. Evaluation of genetic parameters of 23 autosomal STR loci in a southern Chinese Han population., 2018, 45(4): 359–364.
[7] Wang HD, Kand B, Su N, He M, Zhang B, Guo YX, Zhu BF, Liao SX, Zeng ZS. Evaluation of the genetic parameters and mutation analysis of 22 STR loci in the central Chinese Han population,, 2017, 131(1): 103–105.
[8] Qu N, Zhang XC, Liang H, Ou XL. Analysis of genetic polymorphisms and mutations at 23 autosomal STR loci in Guangdong Han population., 2019, 38: e16–e17.
[9] Slooten K, Ricciardi F. Estimation of mutation probabilities for autosomal STR markers., 2013, 7(3): 337–344.
[10] Brinkmann B, Klintschar M, Neuhuber F, Hühne J, Rolf B. Mutation rate in human microsatellites: influence of the structure and length of the tandem repeat., 1998, 62(6): 1408–1415.
[11] Weber JL, Wong C. Mutation of human short tandem repeats., 1993, 2(8): 1123–1128.
[12] Jin B, Su Q, Luo HB, Li YB, Wu J, Yan J, Hou YP, Liang WB, Zhang L. Mutational analysis of 33 autosomal short tandem repeat (STR) loci in southwest Chinese Han population based on trio parentage testing., 2016, 23: 86–90.
[13] Taylor MJ, Fisk NM. Prenatal diagnosis in multiple pregnancy., 2000, 14(4): 663–675.
[14] Wurmb-Schwark NV, Schwark T, Christiansen L, Lorenz D, Oehmichen M. The use of different multiplex PCRs for twin zygosity determination and its application in forensic trace analysis., 2004, 6(2): 125–130.
[15] Xu YJ, Li TT, Pu T, Cao RX, Long F, Chen S, Sun K, Xu R. Copy number variants and exome sequencing analysis in six pairs of Chinese monozygotic twins discordant for congenital heart disease., 2017, 20(6): 521–532.
[16] Weber Lehmann J, Schilling E, Gradl G, Richter DC, Wiehler J, Rolf B. Finding the needle in the haystack: differentiating “identical” twins in paternity testing and forensics by ultra-deep next generation sequencing., 2014, 9: 42–46.
[17] Wang LF, Yang Y, Zhang XN, Quan XL, Wu YM. Tri-allelic pattern of short tandem repeats identifies the murderer among identical twins and suggests an embryonic mutational origin., 2015, 16: 239–245.
[18] Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Magnusson OT, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland., 2017, 549(7673): 519–522.
[19] Valdes AM, Slatkin M, Freimer NB. Allele frequencies at microsatellite loci: the stepwise mutation model revisited., 1993, 133(3): 737–749.
[20] Klintschar M, Dauber EM, Ricci U, Cerri N, Immel UD, Kleiber M, Mayr WR. Haplotype studies support slippage as the mechanism of germline mutations in short tandem repeats., 2010, 25(20): 3344–3348.
[21] Lan Q, Wang HD, Shen CM, Guo YX, Yin CY, Xie T, Fang YT, Zhou YS, Zhu BF. Mutability analysis towards 21 STR loci included in the AGCU 21 + 1 kit in Chinese Han population., 2018, 132(5): 1287–1291.
[22] Ge JY, Budowle B, Aranda XG, Planz JV, Eisenberg AJ, Chakraborty R. Mutation rates at Y chromosome short tandem repeats in Texas populations,, 2009,3 (3): 179–184.
[23] Xu W, Wang YQ, Zhang DD, Wang DX, Zhou L, Ye XL, Zhu CG, Shi YZ. Mutation analysis of 21 autosomal short tandem repeats in Han population from Hunan, China.. 2019, 46(3): 254–260.
[24] Dupuy BM, Stenersen M, Egeland T, Olaisen B. Y-chromosomal microsatellite mutation rates: differences in mutation rate between and within loci,, 2004, 23(2): 117–124.
[25] Xie BB, Chen L, Yang YR, Lv YX, Chen J, Shi Y, Chen C, Zhao HY, Yu ZL, Liu YC, Fang XD, Yan JW. Genetic distribution of 39 STR loci in 1027 unrelated Han individuals from northern China., 2015, 19: 205–206.
[26] Chakraborty R, Stivers DN, Zhong YX. Estimation of mutation rates from parentage exclusion data: applications to STR and VNTR loci., 1996, 354(1):41–48.
[27] Vicard P, Dawid AP. A statistical treatment of biases affecting the estimation of mutation rates., 2004, 547(1–2): 19–33.
[28] Yan JW, Liu YC, Tang H, Zhang QX, Huo ZY, Hu SN, Yu J. Mutations at 17 STR loci in Chinese population., 2006, 162(1–3): 53–54.
[29] Lu DJ, Liu QL, Wu WW, Zhao H. Mutation analysis of 24 short tandem repeats in Chinese Han population., 2012, 126(2): 331–335.
[30] Qian XQ, Yin CY, Ji Q, Li K, Fan HT, Yu YF, Bu FL, Hu LL, Wang JW, Mu HF, Haigh S, Chen F. Mutation rate analysis at 19 autosomal microsatellites., 2015, 36(14): 1633–1639.
Actual mutational research of 19 autosomal STRs based on restricted mutation model and big data
Zhiyong Liu2, He Ren3, Chong Chen4, Jingjing Zhang5, Xiaomeng Zhang1, Yan Shi4, Linyu Shi1, Ying Chen4, Feng Cheng1, Li Jia4, Man Chen6, Qingwei Fan7, Jiarong Zhang1, Wanting Li1, Mengchun Wang1, Zilin Ren8, Yacheng Liu4, Ming Ni8, Hongyu Sun2, Jiangwei Yan1
Short tandem repeat (STR) markers have been widely used in forensic paternity testing and individual identification, but the STR mutation might impact on the forensic result interpretation. Importantly, the STR mutation rate was underestimated due to ignoring the “hidden” mutation phenomenon in most similar studies. Considering this, we useSlooten and Ricciardi’s restricted mutation model based on big data to obtain more accurate mutation rates for each marker. In this paper, the mutations of 20 autosomal STRs loci (and; The restricted model does not include the correction factor of, this paper calculates remaining 19 STR loci mutation rates) were investigated in 28,313 (Total: 78,739 individuals) confirmed parentage-testing cases in Chinese Han population. As a result, total 1665 mutations were found in all loci, including 1614 one-steps, 34 two-steps, 8 three-steps, and 9 nonintegral mutations. The loci-specific average mutation rates ranged from 0.00007700 () to0.00459050 () in trio’s and 0.00000000 () to 0.00344850 () in duo’s. We analyzed the relationship between mutation rates of the apparent and actual, the trio’s and duo’s, the paternal and maternal,respectively. The results demonstrated that the actual mutation rates are more than the apparent mostly, and the values of1*/2*(apparent) are also greater than1/2 (actual) commonly (1*,1;2*,2 are the mutation rates of one-step and two-step). Therefore, the “hidden” mutations are identified. In addition, the mutations rates of trio’s and duo’s, the paternal and maternal, exhibit significant difference. Next, those mutation data are used to do a comparison with the studies of other Han populations in China,which present the temporal and regional disparities. Due to the large sample size, some rare mutation events, such as monozygotic (MZ) mutation and “fake four-step mutation”, are also reported in this study. In conclusion, the estimation values of actual mutations are obtained based on big data, they can not only provide basic data for the Chinese forensic DNA and population genetics databases, but also have important significance for the development of forensic individual identification, paternity testing and genetics research.
autosomal STR; restricted mutation model; parentage testing; Chinese Han population; mutational analysis
2021-06-02;
2021-08-18
国家自然基金重点项目(编号:82030058)资助[Supported by the National Natural Science Foundation of China (No. 82030058)]
刘志勇,博士研究生,研究方向:法医遗传学。E-mail: liuzhy255@mail2.sysu.edu.cn
任贺,硕士,副教授,研究方向:法医遗传学。E-mail: snoopy_fr@sohu.com
刘志勇和任贺并列第一作者。
严江伟,博士,教授,研究方向:法医基因组学。E-mail: yanjw@sxmu.edu.cn
10.16288/j.yczz.21-197
2021/8/31 17:30:44
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20210831.1400.001.html
(责任编委: 朱波峰)
