加权贝叶斯线性B细胞表位特征提取方法
2015-12-29刘威郭红
刘威,郭红
(福州大学数学与计算机科学学院,福建福州 350116)
0 引言
表位是蛋白质抗原性的基础,深入研究蛋白质表位对多肽和新型疫苗分子的设计及诊断试剂的开发具有重要意义[1].线性B细胞表位是抗原序列上能与抗体结合的一段连续的区域[2].早期的表位预测工作主要通过生物实验进行鉴别,但这种方法耗时耗力而且得到的数据较少,针对标准的数据集开发自动评价工具将是未来的发展方向[3].
线性B细胞表位预测问题可以看作一个二分类问题:给定若干肽链序列,指出哪些肽链序列属于表位,哪些属于非表位.表位和非表位在某些生物特性、序列结构、氨基酸组成上存在一定的差异,通过对这些差异提取特征进行分类能够有效地对表位进行预测.B细胞表位数据库的建立,提供了大量的表位序列片段,通过对表位和非表位序列分析,找出表位和非表位的特征,从而进行分类.通常的研究方法是使用一种特征提取方法将肽链序列转化为相应的编码,将编码作为输入再使用分类器进行分类,从而得出预测结果.文献[4]提出了一种氨基酸对抗原性量表(AAPantigenicity scale),并将这种量表作为一种新的编码,该编码反映了表位与非表位中某些特定氨基酸对片段出现频率的差异性,最后使用SVM作为分类器进行预测.文献[5]在AAP编码的基础上提出了长度为3和4的抗原性量表的概念,结合LEP方法,将3种长度的抗原性量表作为分类器的输入参数得出预测模型,这表明AAP编码是一种不错的特征编码.文献[6]中引入贝叶斯中先验概率,提出在线性B细胞表位上的一种贝叶斯特征提取方法,结合氨基酸在肽链序列中的位置形成贝叶斯编码,最后使用SVM进行预测.AAP编码包含了氨基酸对信息,考虑了氨基酸对在表位和非表位数据中的出现频率,但AAP编码对缺少了单个氨基酸的出现频率对肽链序列的影响,这导致肽链信息的缺失.单个氨基酸的贝叶斯编码只包含单个氨基酸与分类结果的关系,没有考虑到氨基酸之间的关系,不符合表位与非表位序列间存在一定结构差异.
受文献[6]的启发,提出一种基于氨基酸对量表加权的贝叶斯特征提取方法,在贝叶斯特征提取方法的基础上,引入氨基酸对抗原性量表的概念,有效地提取肽链序列的组成和结构信息,提高序列与所属类别的相关性.
1 贝叶斯特征提取方法
1.1 贝叶斯特征提取方法的原理
文献[5]中使用贝叶斯方法(Bi-profile Bayes feature extraction)来提取氨基酸序列的特征,并提出贝叶斯特征提取方法的蛋白质甲基化位点预测(BPB-PPM).文献[7]构造了Px用于描述表位和非表位数据中不同氨基酸在不同位置上的差异,将bayes方法应用到线性B细胞表位上.
每条肽链的贝叶斯编码是一个长度2n的向量,其中n表示肽链的长度,每个位置上的氨基酸编码由两个部分构成:对表位数据的先验概率,对非表位数据的先验概率.对2n长度的贝叶斯编码而言,单个贝叶斯编码包含氨基酸在特定位置上出现对类别的先验概率,整体的编码又包含了肽链的氨基酸组成情况.
贝叶斯特征提取方法计算过程描述如下:①将实验数据集分为测试集和训练集;②统计训练集中的每条肽链上单个氨基酸的信息,信息包含肽链序列的不同位置上出现各种氨基酸数量;③使用统计信息构造不同位置上出现氨基酸对肽链类别的先验概率;④使用构造的先验概率对数据集的每一条肽链进行编码,编码时使用氨基酸的种类和位置确定该氨基酸的编码值.
1.2 贝叶斯特征提取方法的不足
贝叶斯特征提取方法以肽链序列与单个氨基酸之间的关系作为特征,然而该方法忽略了氨基酸之间的结构可能对表位和非表位的差异所造成的影响.目前表位和非表位之间存在的明显差异还不得而知,但实际上,表位和非表位存在一定结构上差异,表现为包含数量不同的氨基酸结构体,和结构体在肽链中不同的位置组合.
Bcipep数据库是专门收集B细胞表位数据的数据库,Swiss-Prot数据库是经过注释的蛋白质序列数据库,作为非表位数据库.通过对 Bcipep和Swiss-Prot数据库的氨基酸对组成进行统计,发现表位和非表位的氨基酸对组成有很大差异[4],如图1所示,以每种氨基酸对占总共400氨基酸对的比例作为纵坐标,以氨基酸对FI和MP为例,FI在Bcipep数据库中占所有400种氨基酸对总数的0.067%,MP为0.037%,而在Swiss-Port数据库中这两种氨基酸对占的比例为0.242%和0.111%,通过对比说明表位和非表位数据之间存在氨基酸对的差异,因此很有必要对氨基酸对在表位和非表位数据中的差异信息进行提取.

图1 Bcipep和Swiss-Port数据库中不同氨基酸片段比例Fig.1 Difference of AAPcomposition in Bcipep and Swiss-Port database
2 加权贝叶斯特征提取方法
Bayes编码缺少氨基酸对结构信息,文献[7]中氨基酸对抗原性量的思想是对氨基酸对的一种表示,将其引入到贝叶斯特征提取中,使用滑动窗口将肽链序列分割成氨基酸对,并对氨基酸对进行加权,提出了基于氨基酸对量表加权的贝叶斯特征提取方法.
假设肽链序列S={s1,s2,…,sn},sj(j=1,2,…,n)表示肽链序列中第j个位置的氨基酸s,肽链序列S属于表位或非表位,符号C1表示表位,C-1表示非表位,表1给出三条肽链样本.根据公式有:其中:表示Ci数据中第j个位置上是氨基酸s的概率;P(Ci)表示所有肽链序列数据中Ci数据的概率;P(sj)表示在所有肽链序列数据中第j个位置上是氨基酸s的概率,Psj表示序列S上第j个位置的贝叶斯编码.

表1 三条肽链样本Tab.1 Three peptide chains samples
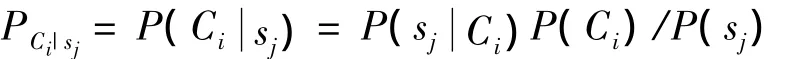

AAP作为一种最简单的氨基酸组合模式和结构,包含了相邻氨基酸相互之间的信息,文献[4]利用同种AAP在表位数据和非表位数据中的频率比值作为该AAP的抗原性量表,使用滑动窗口方式得到AAP编码RAAP,将窗口长度设为2,从头到尾每次向后滑动一个氨基酸,由此可得:

本文提出的改进贝叶斯特征提取方法基于氨基酸对量表加权,采用独立随机变量的方法去除氨基酸对量表中出现的冗余问题.改进后贝叶斯方法不仅包含单个氨基酸与分类结果的关系,还考虑到氨基酸之间的关系,通过增加特征提取所包含的肽链信息提高了预测的准确率.
3 基于改进的贝叶斯特征提取的线性B细胞表位预测方法
使用改进的贝叶斯特征提取方法并结合SVM分类器用于线性B细胞表位的预测,步骤如下.
1)准备数据集,包括表位和非表位数据集,使用El-Manzalawy和Saha数据集,将两组数据集分成训练集和测试集,使用五折交叉验证法生成五组对应的训练集和测试集.
2)特征提取和编码,对两组数据的训练集进行特征提取,使用加权贝叶斯特征提取方法对数据集编码,包括训练集和测试集数据.
3)训练分类器,本文使用SVM作为分类器,使用第二步中已编码的训练集数据作为SVM的输入进行训练,训练过程完成SVM参数的确定.
4)分类器预测,SVM参数确定之后,将第二步中已编码的测试集数据作为分类器的输入,得到分类器对测试集的结果.
5)重复步骤3)、4),使用五组数据中的训练集,并用训练好的分类器测试相应的测试集数据.
4 实验结果与分析
4.1 实验数据集
用于线性B细胞表位预测的标准数据集较多,bcipep数据库[8]是专门收集B细胞表位数据的网上公用数据库,为了更准确地进行测试,本文使用与文献[6]相同的数据集,引用El-Manzalawy和J.Chen已整理的数据集.
1)由El-Manzalawy[9]从bcipep数据库中的947条表位数据中整理的数据集,使用“延伸-截尾”方法得到(30、28、26、24、22、20、18、16、14、12)长度的701条表位数据集.数据下载地址:http://ailab.cs.iastate.edu/bcpreds/.
2)J.Chen等[4]从bcipep数据库中获得并处理得到的固定长度为20的数据,使用“延伸-截尾”方法得到(20、18、16、14、12、10)长度的872条表位数据集.数据下载地址:http://link.springer.com/article/10.1007%2Fs00726-006-0485-9.
El-Manzalawy和S.Saha数据虽然都来自统一数据库,由于处理方式、筛选尺度的不同,数据集仍存在较大的差别,多篇文献也同时引用两个数据集作为试验数据集.
3)从Swiss-Prot数据库中随机生成的固定长度的多肽作为非表位数据.
SVM常用的核函数四种,凭经验选择RBF作为核函数.本文使用的SVM来自于Chang[10]编写的libsvm工具箱.为了提高实验效果,采用libsvm自带的网格搜索算法寻找C和σ2的最优组合.
4.2 评价指标
采用五折交叉验证法,将实验数据分为5个子集,每次把1个子集作为测试集,其余的4个子集作为训练集,每一个子集都被测试过1次,训练过4次,最后将5次结果的平均值作为实际的实验结果.
使用的几个评价指标包括特异性、敏感性、准确率、MCC(Mattew相关系数)、AUC(受试者工作特征曲线下的面积).Rsen反映的是灵敏度,即真实表位被预测为表位的比率;Rspe反映的是特异性,即非表位被预测为非表位的比率;Racc反映的是准确率,即能够被正确预测的表位和非表位的比率;Rpos反映的是阳性预测率,即被预测为表位中真实表位所占的比率;Mattew相关系数是一个性能综合评价指标,其中,真阳性(TP)实际为正样本被正确地预测为正样本的样本数;假阳性(FP)实际为负样本被错误地预测为正样本的样本数;真阴性(TN)实际为负样本被正确地预测为负样本的样本数;假阴性(FN)实际为正样本被错误地预测为负样本的样本数.AUC值为ROC曲线下的面积,AUC越接近于1,说明效果越好.这些参数具体为:
敏感度:Rsen=TP/(TP+FN)×100% 特异度:Rspe=TN/(TN+FP)×100%
精确度:Racc=(TP+TN)/(TP+FP+TN+FN)×100% 阳性预测率:Rpos=TP/(TP+FP)×100%

4.3 实验结果及评价
文献[4]中的贝叶斯特征提取方法对数据提取的贝叶斯编码中包含了训练集和测试集的信息,仅提取训练集数据的信息,对AAP编码也仅提取训练集数据信息.在两个数据集上将加权贝叶斯特征提取方法,与贝叶斯特征提取方法作对比.
表2、3是在El-Manzalawy数据集上使用贝叶斯特征提取方法,加权贝叶斯方法在不同长度窗口(12、14、16、18、20、22、24、26、28、30)下的参数,由于都是采用SVM训练分类器来进行分类,因此分类效果的区别主要来自特征提取方法,可以看出改进的贝叶斯方法除了特异度以外,在其他指标上整体参数均有提升.当窗口长度大于20时提升更加明显,在窗口长度为26、28、30时准确率的提升达到了10个百分点.需要强调的是目前90%的表位数据长度都在20以下,数据集中长度大于20的表位数据都是利用源数据扩展得来,但是在一定程度上也表现出数据的性质,因此采用了大于20长度的表位数据集(22、24、26、28、30).
表4、5是在Saha数据集上对贝叶斯特征提取方法和加权贝叶斯特征提取方法的比较,从表中可以看出,在Saha数据集上,各个长度的数据的实验结构相比贝叶斯方法改进后有较大的提升.20为肽链的经典长度,在该长度下将两个数据集的两种特征提取方法绘制ROC曲线(图2).从图2可以看出,在El-Manzalawy数据集上,由于改进前方法的特异度高于改进之后的特异度值,两条ROC曲线的AUC值并无太大差异;在Saha数据集上,改进后方法的敏感度和特异度均优于改进前方法,可以看出ROC曲线下的AUC值有明显提升.

表2 贝叶斯特征提取在El-Manzalawy数据集上的参数Tab.2 Using Bayes feature extraction in El-Manzalawy dataset
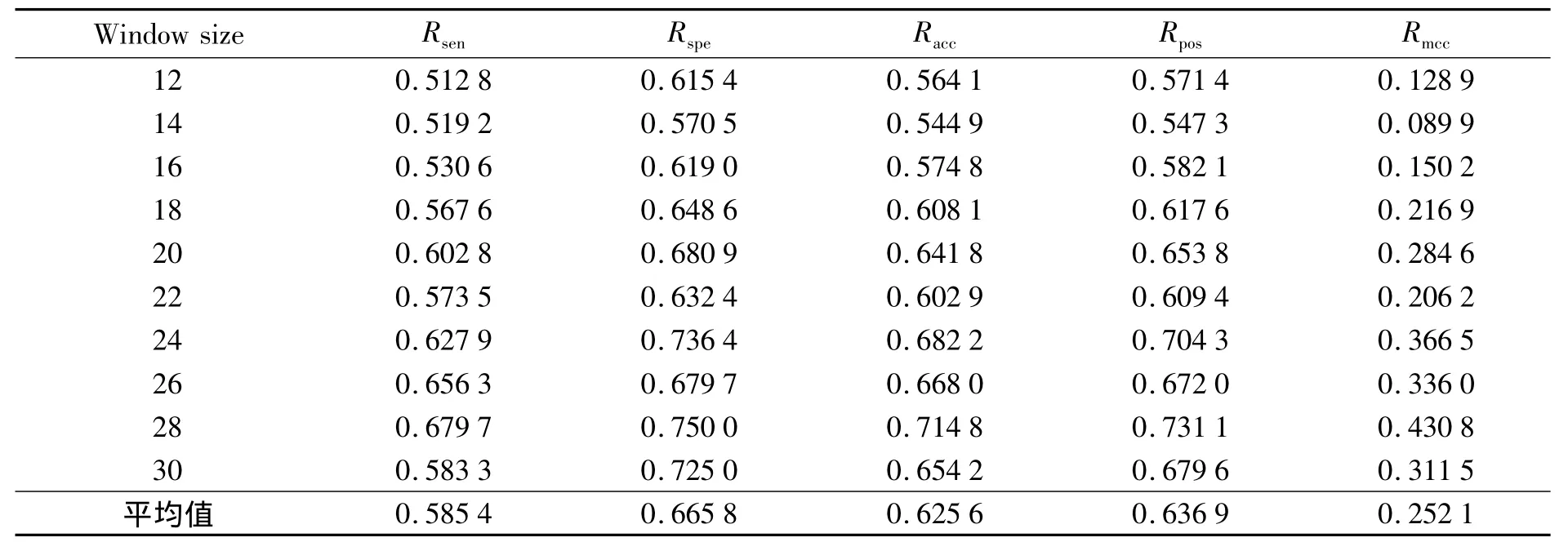
表3 加权贝叶斯特征提取在El-Manzalawy数据集上的参数Tab.3 Using weighted Bayes feature extraction in El-Manzalawy dataset

表4 贝叶斯特征提取在Saha数据集上的参数Tab.4 Using Bayes feature extraction in Saha dataset

表5 加权贝叶斯特征提取在Saha数据集上的参数Tab.5 Using weighted Bayes feature extraction in Saha dataset
目前预测线性B细胞表位的方法较多,所提取的特征也不尽相同,但总体预测效果差不多.AAP方法是一种常用的线性B细胞表位预测方法,该方法以氨基酸对作为特征提取的基本单位.为了进一步说明本文提出方法的有效性,下面给出本文提出方法与AAP方法的对比实验,为了避免数据人工处理带来的误差,这里取长度为20的表位数据在前面所述两个数据集上进行试验比对.

图2 El-Manzalawy,Saha数据集的两种贝叶斯特征提取方法ROC曲线Fig.2 Both bayes and weighted bayes feature extraction ROC on El-Manzalawy,Saha dataset
通过两个数据集的实验结果可以看出,AAP方法的敏感度(Rsen)比贝叶斯方法高,这说明此类特征能更准确描述表位数据,而加权贝叶斯方法中的敏感度高于两种方法,表明加权贝叶斯方法在描述表位数据上更加有效;特异度(Rspe)一定程度上受到敏感度的影响,使得加权贝叶斯方法在特异度上数值有所降低,由于表位数据来源于真实数据,而非表位数据是从数据库中随机生成,这也影响了非表位数据的精度和实验结果;准确率反映了整体的预测精度,加权贝叶斯方法相比其他两种方法表现出了较好精度.从表6、7的结果可知,相比AAP算法,加权贝叶斯算法有更好的性能效果.

表6 贝叶斯,加权贝叶斯,AAP提取方式在El-Manzalawy数据集上的参数Tab.6 Using Bayes,w -Bayes,AAP feature extraction in El-Manzalawy dataset

表7 贝叶斯,加权贝叶斯,AAP提取方式在Saha数据集上的参数Tab.7 Using Bayes,w -Bayes,AAP feature extraction in Saha dataset
5 结论
对线性B细胞表位的预测是一个重要的问题,而特征提取是问题的重心所在.针对特征提取提出了一种基于氨基酸对量表加权的贝叶斯特征提取方法.在对氨基酸序列的特征提取上考虑了结构的特征,把一种AAP量表引入到贝叶斯特征提取上来,根据对El-Manzalawy和Saha数据集的实验,该方法能够提高贝叶斯特征提取方法的预测精度.今后的研究工作重点是如何将改进贝叶斯的特征提取方法应用到线性B细胞表位的预测中去.
