反式双氧锰(V)咔咯配合物的稳定性
2015-12-29章小慧徐志广龚丽珍沈桂贤陈华彬刘海洋
章小慧 徐志广 龚丽珍 许 旋 沈桂贤 陈华彬 刘海洋
(1华南师范大学化学与环境学院,教育部环境理论化学重点实验室,广州510006;2华南理工大学化学系,广州510641)
反式双氧锰(V)咔咯配合物的稳定性
章小慧1徐志广1,*龚丽珍1许 旋1沈桂贤1陈华彬1刘海洋2,*
(1华南师范大学化学与环境学院,教育部环境理论化学重点实验室,广州510006;2华南理工大学化学系,广州510641)
采用密度泛函理论(DFT)的B3LYP方法对反式双氧锰(V)咔咯配合物阴离子的稳定性及其质子化物种进行了理论计算.结果表明:反式双氧锰(V)咔咯配合物阴离子构型稳定,其反式双氧锰键O=Mn=O由锰原子的d轨道与两个氧原子的p轨道分别构成一个σ轨道和两个π轨道;随着外围取代基吸电性增强,O=Mn=O键长缩短,拉曼伸缩振动频率增大;其质子化过程中得到两个质子的轴向氧原子与锰原子的距离超出正常化学键的范围,从而形成水分子并脱离原来分子,导致质子化行为是不可逆过程,而形成单氧的咔咯锰(V)-氧配合物.
密度泛函理论;反式双氧锰(V)咔咯配合物;稳定性;质子化
1 引言
过渡金属-氧配合物在催化氧化、生物化学和某些无机反应中起着相当重要的作用.1-3在光合系统II中,锰簇化合物是水转化成氧气关键的催化中间体.4-6在模拟光合系统产氧的过程7-9中研究最多的化合物是锰卟啉类配合物.1997年Groves等10获得锰(V)-氧卟啉化合物,该化合物既能参与氧转移反应也可用作模拟生物酶的催化氧化反应.在进一步对锰(V)-氧卟啉化合物催化氧化反应实验研究中发现该过程是涉及3个质子化产物的酸碱平衡系统,并首次获得重要中间产物——具反式结构的双氧锰(V)卟啉配合物[MnVO2(por)]-.11,12双氧锰卟啉配合物能在酸性条件下分步质子化为OMnIV(OH)(por)和[OMnIII(OH2)(por)]+,并能随酸碱条件不同而相互转化.对上述配合物[OMnX(por)]+(X=H2O,OH-,O2-)的理论计算结果显示体系的构型是稳定的,其轴向氧基作用的影响大小为O2->OH->H2O.13
咔咯是具有18π电子结构的类卟啉大环化合物,其中有两个吡咯环直接相连,易于形成三价阴离子配体,具有空腔小的特点.金属咔咯配合物的催化氧化性能优于金属卟啉配合物,14锰咔咯配合物在氧化剂的条件下,会催化有机底物发生氧化反应伴随氧转移反应.15-18因而咔咯配合物成为卟啉类化合物的研究热点.与锰卟啉配合物的反应性质不同,锰咔咯配合物在类似的催化氧化反应中,至今未发现生成相应的反式双氧锰(V)咔咯配合物.8因此反式双氧锰(V)咔咯配合物及其质子化产物能否稳定存在,至今尚未有相关研究报道.
针对上述反式双氧锰(V)咔咯配合物稳定性的问题,本文运用密度泛函理论(DFT)方法对反式双氧锰(V)咔咯配合物[1-OMnO]2-及其质子化结构[1-OMnX](X=OH-,H2O)(图1)进行理论计算,研究体系的稳定性以及取代基的影响.从理论上证明反式双氧锰(V)咔咯配合物及其质子化产物能稳定存在,为后续的理论和实验研究提供新的理论依据.

图1 反式双氧锰(V)咔咯配合物及其质子化物的分子结构Fig.1 M olecular structuresof trans-dioxo M n(V)corrole com p lex and protonation species

表1 单重态下DFT方法优化的[1-OM nO]2-的部分结构参数Table 1 Some optim ized structuralparam etersof[1-OM nO]2-w ith DFTmethod for the singlet state
2 计算方法
为优选计算方法,以无取代基的反式双氧锰(V)咔咯[1-OMnO]2-结构为标准,选用BP86、19,20PBEPBE、21,22B3LYP23三种常见DFT方法,Mn原子采用LanL2DZ基组,C、H、N、F、O用6-31G(d,p)基组进行优化计算,优化的主要几何数据列于表1.与反式双氧锰卟啉(V)配合物相比,发现各方法优化的键长dMn=O均在合理的键长范围内,11其中B3LYP方法计算得出的dMn=O更短.我们课题组24以[Br8TPFC]Mn(EtOAc)(TPFC:5,10,15-三(五氟苯基)咔咯)单晶结构为标准为咔咯锰(V)配合物优选计算方法时,发现选用B3LYP方法优化的结构尤其是dMn≡O更接近实验值,因此本文采用B3LYP方法,Mn原子用LanL2DZ基组,C、H、N、F、O用6-31G(d,p)基组对系列分子进行优化和频率验证.
本文在反式双氧锰(V)咔咯配合物的meso-和β-位引入不同的取代基,设计了四种带不同取代基的分子(图1),配合物meso-和β-位取代基按拉电子能力由弱到强的顺序依次为H/H、C6H5/H、C6F5/H和C6F5/F,分别记为[1-OMnO]2-、[2-OMnO]2-、[3-OMnO]2-和[4-OMnO]2-.参考文献13卟啉配合物的计算方案,同时为考察多重态的计算结果并进行对比,采用B3LYP的方法对各配合物单、三、五重态下的结构进行优化计算,并计算其振动频率,进行频率验证.为探究反式双氧锰(V)咔咯配合物质子化过程的稳定性,对其质子化的结构进行计算分析.因反式双氧锰(V)卟啉配合物中锰原子的化合价为+5价,25-27故本文配合物中锰原子的化合价设为+5价.并对优化的几何构型进行自然键轨道(NBO)理论28计算,所有的计算均采用Gaussian 03程序包.29
3 结果与讨论
3.1 O=Mn=O成键及其稳定性
对反式双氧锰(V)咔咯[1-OMnO]2-单、三、五重态下的结构进行几何优化,频率验证显示均无虚频,表示体系构型稳定.单重态下优化的结构如图2所示,优化的能量和部分几何结构参数见表2,能量高低顺序为三重态>五重态>单重态,说明[1-OMnO]2-在单重态下最稳定.三、五重态下优化的结构与单重态相似,呈C2v对称结构.由于咔咯的两个吡咯环直接相连,[1-OMnO]2-的对称性比相应的反式双氧锰(V)卟啉[MnV(O)2TPFPP]-11的D4h对称性要低.[1-OMnO]2-的∠O5MnO6在单、三、五重态下的数值都为176º左右,接近180°,说明O5、Mn、O6三个原子近似共线,∠O6MnN1N2的数值接近90°,∠MnN2N3N4的数值为0°,表明Mn处在大环的平面上,且O5、Mn、O6三个原子形成的直线与大环平面垂直.dMn=O5与dMn=O6的数值相等,说明Mn=O5和Mn=O6键型是相同的,单、三重态下的dMn―O近似相等,而五重态下的键长拉长了0.017 nm,说明结构受自旋态的影响较大,更高自旋态五重态下的Mn=O键可能更容易断裂,易发生氧转移反应.

图2 单重态下[1-OMnO]2-优化后的几何结构Fig.2 Optim ized structureof[1-OM nO]2-for the singlet state

表2 不同自旋态下[1-OM nO]2-优化的能量和部分几何结构参数Table2 Optim ized energy and selected structural parametersof[1-OMnO]2-in different spinmultipilicities
对反式双氧锰(V)咔咯配合物进行电子组态和轨道分析,各自旋态的电子组态图见图3,单重态下锰原子的dxy、dxz、dyz和dz2轨道参与成键,而dx2-y2没有参与成键,为非键轨道,被两个电子占据,锰原子的与两个氧原子的pz轨道作用形成一个σ轨道,锰原子的dxz、dyz分别与两个氧原子的px和py形成πxz和πyz轨道,轨道分析显示[1-OMnO]2-的锰原子与氧原子存在成键作用,同时锰原子的dxy与吡咯的N形成σ轨道,相比于对应的卟啉锰(V)氧配合物,[1-OMnO]2-多出了πpx-px和πpy-py轨道(见图3).前线轨道附近,存在与卟啉的a1u和a2u类似的轨道,30记为a1和a2轨道,主要电子云分布在四个吡咯上的N和C原子上.[1-OMnO]2-的价电子组态为(a1)2(a2)2,同时分析[1-OMnO]2-三重态和五重态下的价电子组态进行对比,三重态下的价电子组态为(πxz)2(πyz)2(dx2-y2)2(πpx-px)2(πpy-py)2(a1)2(a2)1(πcor)1,从电子组态图可以看出a2轨道上的一个电子跃迁到分布在咔咯骨架上的另一个π轨道上,其余特征轨道的能量和电子没有明显变化,因此三重态下的两个dMn=O与单重态下的几乎相等.五重态下的电子组态为(πxz)2(πyz)2(dx2-y2)1(πpx-px)2(πpy-py)1(a1)2(a2)1(π*xz)1(π*yz)1,和πpy-py轨道分别有一个电子跃迁到π*xz和π*yz轨道上,Mn=O的键级减少,键长拉长到0.1806 nm,键的强度有所减弱,解释了高自旋态下配合物更具反应活性的实验现象.13
为了更深入讨论O5、Mn、O6三个原子之间的键合作用,对单重态[1-OMnO]2-的O=Mn=O键进行NBO分析,结果表明,锰原子与氧原子间形成了一个双电子占据的σ键和两个双电子占据的π键及相应的空反键轨道(见图4).由于体系为C2v对称,两个氧原子等同,说明O6、Mn、O5三个原子之间确实形成有效的化学键,[1-OMnO]2-体系中反式锰双氧键O=Mn=O较稳定.

图3 [1-OM nO]2-在单(S),三(T),五重态(Q)下的分子轨道Fig.3 M olecular orbitals for the singlet(S),triplet(T),and quintet(Q)statesof[1-OMnO]2-
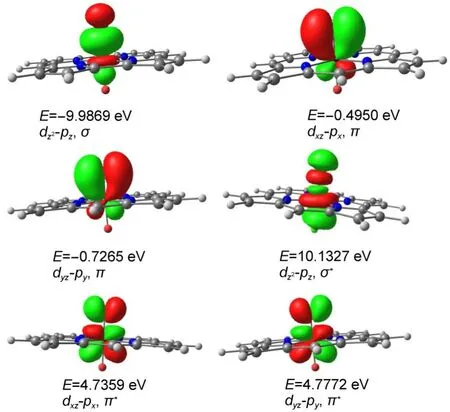
图4 单重态[1-OM nO]2-的M n=O键的自然键轨道Fig.4 Naturalbond orbital(NBO)ofM n=O bond of [1-OM nO]2-in singlet state

表3 [n-OM nO]2-(n=1,2,3,4)的部分几何结构参数Table 3 Selected structure parametersof[n-OM nO]2-(n=1,2,3,4)
3.2 取代基对反式双氧锰(V)咔咯结构的影响
本课题组以往的工作发现具有推拉电子作用的外围取代基会改变dMn=O,影响配合物的氧转移能力,24因此本文考虑在反式双氧锰咔咯配合物的meso-和β-位引入不同拉电子能力的取代基(C6H5, C6F5和F),探究取代基效应对反式双氧锰(V)咔咯配合物结构特别是dMn=O的影响,综合meso-和β-位取代基拉电子能力大小按下列配合物排列依次增大排序: [1-OMnO]2->[2-OMnO]2->[3-OMnO]2->[4-OMnO]2-.
上述配合物在不同自旋态下进行优化,优化的结构参数见表3,表3显示在单、三重态下,随着取代基拉电子能力增大,配合物O=Mn=O键的键长呈规律性缩短,单重态下随着取代基吸电子能力增强,dMn=O5依次为0.1638、0.1636、0.1635和0.1633 nm.dMn=O6依次为0.1638、0.1637、0.1636和0.1634 nm.说明吸电性取代基会增强O=Mn=O键的成键作用,从结构数据看出不同取代基的影响下Mn仍保持在大环平面,并且与双氧和大环形成的角度均没有发生改变,说明单、三重态下外围取代基对配合物的构型影响不大.五重态下随取代基吸电性增强,dMn=O不再呈规律性变化.考虑到Mn的价电子组态为3d54s2,显示Mn为+5时,很难形成稳定的4个单电子的五重态.所以推断配合物的五重态可能为不稳定态.dMn=O呈不规律变化的原因可能是不稳定的五重态对反式双氧锰咔咯配合物的结构的干扰所造成的.

图5 [n-OM nO]2-(n=1,2,3,4)中O=M n=O的拉曼对称伸缩振动频率Fig.5 Raman symmetricalstretching frequencies of O=Mn=O of[n-OMnO]2-
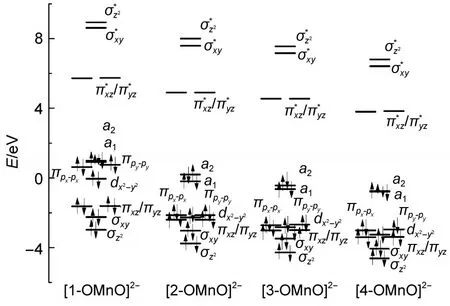
图6 [n-OM nO]2-(n=1,2,3,4)单重态下的分子轨道分布Fig.6 M olecular orbitaldistributions for the singlet stateof[n-OMnO]2-(n=1,2,3,4)

图7 单重态[1-OMnO]2-的前线轨道Fig.7 Frontiermolecular orbitals for the singletstateof [1-OM nO]2-
Mn=O键的伸缩振动可在拉曼光谱中呈现明显的吸收峰,因此配合物中Mn=O键合程度可以通过拉曼光谱来测定表征.Corrolazine MnVO的Mn≡O键的振动吸收峰在970-981 cm-1范围内,31认为其Mn≡O是一个三键结构,且Mn≡O的振动频率会受取代基影响.反式双氧锰(V)卟啉配合物的Mn=O振动频率在741-744 cm-1范围内,该Mn=O是一个双键结构.11说明dMn=O的振动频率越大键级越高,本课题组通过理论计算方法研究表明咔咯锰(V)-氧配合物Mn≡O是三键结构,计算的拉曼振动吸收峰校正后在999-1036 cm-1范围内,计算结果说明吸电性取代基会增强Mn≡O键的强度,Mn≡O键长缩短,频率νMn=O往高频移动.24Jin等11也发现Mn≡O之间的键越强,振动频率越大.本文计算单重态下的[n-OMnO]2-(n=1,2,3,4)的拉曼振动吸收峰.经校正后在828-845 cm-1范围内,介于三键和双键的振动范围之间(见图5).外围取代基为H的[1-OMnO]2-,O=Mn=O的振动频率为828.41 cm-1,随着外围取代基的吸电性增强,相应的O=Mn=O振动频率增大,吸电性最强的[4-OMnO]2-的O=Mn=O振动频率最大,为844.12 cm-1,说明取代基对[n-OMnO]2-的O=Mn=O的振动频率有一定影响,规律与咔咯锰(V)-氧配合物一致.24
为了更深入地探讨取代基效应的内在因素,对电子组态和轨道进行分析,单重态下[n-OMnO]2-(n=1,2,3,4)的电子组态图见图6,从图可以看出,外围取代基并没有影响[n-OMnO]2-的电子组态排布,电子组态都为(πxz)2(πyz)2(dx2-y2)2(πpx-px)2(πpy-py)2(a1)2(a2)2,但是它们的轨道能级发生变化,特征能级相应下降,其σz2
轨道的能级下降,依次为-2.97、-3.76、-4.28和-4.62 eV,说明环外围连有吸电基会降低成键轨道能级,导致O=Mn=O成键作用增强.
上述配合物HOMO和LUMO轨道分布类似, [1-OMnO]2-的前线轨道分布见图7,前线轨道的电子云主要分布在咔咯骨架上.同时分析了前线轨道的能级和相应能隙(见图8).[1-OMnO]2-的HOMO和LUMO的能级分别为2.59和4.87 eV,当中位连有苯基时,HOMO和LUMO能级分别降低到1.71和3.77 eV,连有吸电性最强取代基的[4-OMnO]2-的HOMO和LUMO能级分别降低到0.42和2.74 eV.说明外围取代基的吸电性增强会明显降低前线轨道能级,但对能隙变化影响不大.

图8 [n-OMnO]2-(n=1,2,3,4)单重态下前线轨道能级及能隙图Fig.8 Energiesand energy gapsof frontier orbitals of[n-OM nO]2-(n=1,2,3,4)atsinglet state

图9 单,三,五重态下[1-OM nOH]-和[1-OM nH2O]优化的几何结构Fig.9 Optim ized structure of[1-OM nOH]-and [1-OM nH2O]at singlet,trip let,and quintet states

表4 不同自旋态下分子[1-OMnX](X=H2O,OH-)的能量和部分几何结构参数Table 4 Energiesand selected structure parametersof[1-OMnX](X=H2O,OH-)in different spinmultipilicities
3.3 质子化过程
对锰(V)氧卟啉配合物研究发现它的催化氧化过程是涉及多个质子化产物的酸碱平衡系统,产物随酸碱条件不同而相互转化,能生成稳定的反式双氧锰(V)卟啉配合物,而且发现质子化产物的氧转移活性和反应性更好.13锰(V)氧咔咯配合物在此类催化氧化反应中不生成对称的反式双氧锰(V)咔咯配合物,而是在咔咯环上同一侧两个氧原子直接相连生成环状的过氧化合物,32本文对反式双氧锰(V)咔咯配合物质子化过程进行计算,试图探究质子化过程中各配合物的结构和稳定性.参考反式双氧锰(V)氧卟啉配合物的质子化过程,13[1-OMnO]2-质子化过程中分别形成化合物[1-OMnOH]-和[1-OMnH2O],分别对其质子化的结构在单、三、五重态进行优化.各自旋态下各配合物优化的结构见图9,优化的能量和几何结构参数见表4,频率验证均无虚频,计算结果显示,分子[1-OMnOH]-在单、三、五重态优化的能量相近,优化的质子化结构不具有对称面和对称轴,dMn=O5与dMn=O6的数值产生显著差别,单重态dMn=O5增至0.1889 nm,dMn=O6缩短至0.1565 nm,相差约0.03 nm左右.∠O5MnO6为166.1°,三个原子偏离同一条直线,∠MnN2N3N4为-6.2°,Mn原子偏离大环平面.三重态下[1-OMnOH]-中∠O5MnO6接近180°,∠MnN2N3N4为-6.0°,相比单重态dMn=O5和dMn=O6都相应增长0.01 nm左右.五重态下[1-OMnOH]-的结构与单重态下相差较大,∠O5MnO6接近180°,∠MnN2N3N4为-0.2°,Mn基本在大环平面上,dMn=O5与dMn=O6的差值明显变小,仅为0.003 nm左右,dMn=O5和dMn=O6的强度很接近.上述说明质子化的结构[1-OMnOH]-中氢原子的引入会改变反式双氧锰(V)咔咯配合物的构型,使得dMn=O5键长增长,相应的dMn=O6缩短,且[1-OMnOH]-构型受自旋多重态影响较大.
[1-OMnH2O]的最稳定态为单重态,单重态下分子[1-OMnH2O]中dMn=O5增至0.3587 nm,Mn与O5距离超出正常化学键的范围,说明Mn与O5已不再形成有效化学键,且dMn=O6的数值(0.1539 nm)与单重态下的(TPFC)MnO的Mn≡O键长(0.1548 nm)24很接近,说明[1-OMnH2O]分子中Mn≡O6之间是一个三键结构.三重态下[1-OMnH2O]构型与单重态类似,dMn=O5为0.3187 nm,dMn=O6为0.1575 nm,Mn原子同样明显偏离大环平面为-21.5°,五重态下的[1-OMnH2O]中dMn=O5的数值明显减小为0.24 nm左右,键长仍大于反式双氧卟啉的相应键长(约0.22 nm),11显示Mn=O5键间作用较弱;dMn=O6的数值增至0.1645 nm,说明分子[1-OMnH2O]构型同样受自旋多重态影响较大,这与Balcells等13报道过的反式双氧锰(V)卟啉的氧转移反应速率受自旋态能隙影响的规律是类似的.受到Mn≡O6键合程度的影响,∠MnN2N3N4在五重态的数值为-10.3°,相比于单重态下的-22.9°,三重态下的-21.5°说明Mn偏离的程度明显减小.
根据以上讨论,单重态下反式双氧锰(V)咔咯配合物的质子化过程可由图10表示.其质子化结构均能稳定存在,质子化过程中随着反式双氧锰(V)咔咯配合物得到质子,轴向氧基配体与锰原子形成的键长逐渐拉长.单重态下配合物在得到一个质子的过程a中形成配合物[1-OMnOH]-,dMn=O5有所削弱,键长与反式双氧锰(V)卟啉配合物相近,过程应可逆.在质子化过程b中,[1-OMnH2O]结构中O5得到两个质子后形成水分子,dMn=O5超出正常化学键的范围,因此可认为,与反式双氧锰(V)卟啉配合物的可逆质子化过程不同,反式双氧锰(V)咔咯配合物质子化b过程应是不可逆的过程.这过程致使轴向O5质子化形成水分子脱离原来分子,并最终生成咔咯锰(V)-氧配合物.据此,理论上尽管[1-OMnO]2-构型稳定,但是因为不可逆质子化b过程的存在,而可能导致[1-OMnO]2-在催化氧化过程中被质子化为咔咯锰(V)-氧配合物而难以被检测到出来.

图10 单重态下反式双氧锰(V)咔咯配合物的质子化过程Fig.10 trans-Dioxomanganese corrole(V)protonated processat singlet state
4 结论
采用B3LYP方法对meso-和β-位连有不同取代基的反式双氧锰(V)咔咯配合物及其质子化过程进行理论计算研究.结果显示,反式双氧锰(V)咔咯配合物及其质子化产物均能稳定存在.反式双氧锰键O=Mn=O由一个σ轨道和两个π轨道构成,外围取代基效应对双氧锰(V)咔咯配合物中O=Mn=O键的影响呈规律性变化,随着取代基拉电子能力的增强dMn=O相应缩短,相应的拉曼振动频率向高频移动,电子组态分析结果显示取代基效应并没有改变配合物的电子组态排布.考察反式双氧锰(V)咔咯配合物的质子化过程,发现引入两个质子后,轴向氧原子会质子化形成水分子而脱离原来分子,导致质子化过程不可逆,反式双氧锰(V)咔咯配合物最终转化成咔咯锰(V)-氧配合物.
Suppo rting In fo rm ation:The stability of anionic transdioxo manganese(V)corrole complex and the protonated species structure were investigated using theoretical density functional theory(DFT)w ith B3LYPmethod.This information is available free of charge via the internet at http://www.whxb. pku.edu.cn.
(1)Reedijk,J.Bioinorganic Catalysis;Reedijk,J.Ed.;Marcel Dekker Inc.:New York,1993;pp 347-393.
(2)Meunier,B.Biomimetic OxidationsCatalyzed by Transition MetalComplexes;Meunier,B.Ed.;ImperialCollege Press: London,2000.
(3)Sheldon,R.A.Metalloporphyrins in Catalytic Oxidations; Sheldon,R.A.Ed.;MarcelDekker Inc.:New York,1994.
(4)M cEvoy,J.P.;Brudvig,G.W.Chem.Rev.2006,106,4455.doi: 10.1021/cr0204294
(5)Yagi,M.;Kanako,M.Chem.Rev.2001,101,21.doi:10.1021/ cr980108l
(6)Rebelo,S.L.H.;Pereira,M.M.;Simoes,M.M.Q.;Neves,M. G.;Cavaleiro,J.A.S.J.Catal.2005,234,76.doi:10.1016/j. jcat.2005.05.026
(7)Shimazaki,Y.;Nagano,T.;Takesue,H.;Ye,B.H.;Tani,F.; Naruta,Y.Angew.Chem.Int.Edit.2004,43,98.
(8)Gao,Y.;Åkermark,T.;Liu,J.H.;Sun,L.C.;Akermark,B. J.Am.Chem.Soc.2009,31,8726.
(9)Liu,X.;Wang,F.Y.Coord.Chem.Rev.2012,256,1115.
(10)Groves,J.T.;Lee,J.;Marla,S.S.J.Am.Chem.Soc.1997,119, 6269.doi:10.1021/ja962605u
(11)Jin,N.;Ibrahim,M.;Spiro,T.G.;Groves,J.T.J.Am.Chem. Soc.2007,129,12416.doi:10.1021/ja0761737
(12)Gross,Z.Angew.Chem.Int.Edit.2008,47,2737.
(13)Balcells,D.;Raynaud,C.;Crabtree,R.H.;Eisenstein,O.Inorg. Chem.2008,47,10090.doi:10.1021/ic8013706
(14)Gross,Z.;Simkhovich,L.;Galili,N.Chem.Commun.1999,599.
(15)Gross,Z.;Golubkov,G.;Simkhovich,L.Angew.Chem.Int. Edit.2000,39,4045.
(16)Kumar,A.;Goldberg,I.;Botoshansky,M.;Buchman,Y.;Gross, Z.J.Am.Chem.Soc.2010,132,15233.doi:10.1021/ja1050296
(17)Liu,H.Y.;Mahmood,M.H.R.;Qiu,S.X.S.;Chang,C.K. Coord.Chem.Rev.2013,257,1306.doi:10.1016/j. ccr.2012.12.017
(18)Aviv,I.;Gross,Z.Chem.Commun.2007,1987.
(19)Becke,A.D.Phys.Rev.A 1988,38,3098.doi:10.1103/ PhysRevA.38.3098
(20)Perdew,J.P.Phys.Rev.B 1986,33,8822.doi:10.1103/ PhysRevB.33.8822
(21)Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.Lett.1997, 78,1396.
(22)Perdew,J.P.;Burke,K.;Ernzerhof,M.Phys.Rev.Lett.1996, 77,3865.doi:10.1103/PhysRevLett.77.3865
(23)Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/ 1.464913
(24)He,J.;Xu,Z.G.;Zeng,Y.X.;Xu,X.;Yu,L.;Wang,Q.;Liu,H. Y.Acta Phys.-Chim.Sin.2012,28,1658.[何 婧,徐志广,曾允秀,许 旋,喻 兰,王 琦,刘海洋,物理化学学报,2012, 28,1658.]doi:10.3866/PKU.WHXB201205101
(25)DeAngelis,F.;Jin,N.;Car,R.;Groves,J.T.Inorg.Chem.2006, 45,4268.doi:10.1021/ic060306s
(26)Jin,N.;Lahaye,D.E.;Groves,J.T.Inorg.Chem.2010,49, 11516.doi:10.1021/ic1015274
(27)Arunkumar,C.;Lee,Y.M.;Lee,J.Y.;Fukuzumi,S.;Nam,W. Chem.-Eur.J.2009,15,11482.doi:10.1002/chem.200901362
(28)Reed,A.E.;Curtiss,L.A.;Weinhold,F.Chem.Rev.1988,88, 899.
(29)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;etal.Gaussian 03, Revision A.01;Gaussian Inc.:Pittsburgh,PA,2003.
(30)Gouterman M.J.Mol.Spectrosc.1961,6,138.doi:10.1016/ 0022-2852(61)90236-3
(31)Mandimutsira,B.S.;Ramdhanie,B.;Todd,R.C.;Wang,H.; Zareba,A.A.;Czernuszew icz,R.S.;Goldberg,D.P.J.Am. Chem.Soc.2002,124,15170.doi:10.1021/ja028651d
(32)Kim,S.H.;Park,H.;Seo,M.S.;Kubo,M.;Ogura,T.;Klajn,J.; Gryko,D.T.;Valentine,J.S.;Nam,W.J.Am.Chem.Soc.2010, 132,14030.doi:10.1021/ja1066465
Stability o f trans-Dioxo Manganese(V)Co rro le Com p lex
ZHANG Xiao-Hui1XU Zhi-Guang1,*GONG Li-Zhen1XU Xuan1SHEN Gui-Xian1CHEN Hua-Bin1LIU Hai-Yang2,*
(1SchoolofChemistry and Environment,Key Laboratory ofTheoreticalChemistry ofEnvironment, Ministry ofEducation,South China NormalUniversity,Guangzhou 510006,P.R.China;2DepartmentofChemistry,South China University ofTechnology,Guangzhou 510641,P.R.China)
The stability ofanionic trans-dioxo m anganese(V)corrole com p lex and the protonated species structure were investigated using density functional theory(DFT)w ith B3LYPmethod.The calculation results show that trans-dioxomanganese(V)corrole com plex has oneσand twoπorbitals in its O=Mn=O bonds, which are com posed of the d orbital of the manganese atom and p orbitals of the two oxygen atom s. Enhancementof the electron-w ithdraw ing ability ofsubstituents results in a decrease in the O=Mn=O bond lengths,and shifts the O=Mn=O Raman stretching vibration to a higherwavenumber.On protonation,one of the axialoxygen atoms gains two protons and is transformed into a watermolecule.Themanganese atom then cannot hold water tightly to form effective coordination bonds w ith water.This results in irreversible protonation of the trans-dioxomanganese(V)corrole com p lex,which leads to formation ofan oxomanganese (V)corrole com plex.
Density functional theory;trans-Dioxomanganese(V)corrole comp lex;Stability; Protonation
O641
icle]
10.3866/PKU.WHXB201504022 www.whxb.pku.edu.cn
Received:February 2,2015;Revised:April1,2015;Published onWeb:April2,2015.
∗Corresponding authors.XU Zhi-Guang,Email:chzgxu@scnu.edu.cn;Tel:+86-20-39310187.LIU Hai-Yang,Email:chhyliu@scut.edu.cn; Tel:+86-20-22236805.
The projectwassupported by the NationalNatural Science Foundation of China(21171057,21275057,21371059)and Natural Science Foundation ofGuangdong Province,China(S2012010008763).
国家自然科学基金(21171057,21275057,21371059)和广东省自然科学基金(S2012010008763)资助项目
©Editorialofficeof Acta Physico-Chim ica Sinica