抗癌药物丙卡巴肼的合成工艺再优化
2015-12-24汪亚帷刘笑天陈河如
潘 龙,汪亚帷,刘笑天,陈河如,2
(1.暨南大学药学院中药及天然药物研究所,广东广州510632;2.广东省中药药效物质基础及创新药物研究重点实验室,广东广州510632)
抗癌药物丙卡巴肼的合成工艺再优化
潘 龙1,汪亚帷1,刘笑天1,陈河如1,2
(1.暨南大学药学院中药及天然药物研究所,广东广州510632;2.广东省中药药效物质基础及创新药物研究重点实验室,广东广州510632)
以对甲基苯甲酸为起始原料,与氯化亚砜反应得对甲基苯甲酰氯(2),酰氯直接与异丙胺反应得对甲苯甲酰异丙胺(3);化合物3经硝酸铈铵(CAN)氧化得4-甲酰基苯甲酰异丙胺(4);以氰基硼氢化钠为还原剂,化合物4与甲基肼硫酸盐在碱性条件下经经缩合还原反应生成目标化合物丙卡巴肼.该工艺路线共4步反应,总收率44.7%,每个单元反应收率71%~86%.所有化合物的结构均经1H-NMR、13C-NMR和MS确认.
丙卡巴肼;合成工艺;药物合成;抗肿瘤
丙卡巴肼(procarbazine),化学名为N-异丙基-(2-甲基肼基)对甲苯甲酰胺,也称甲苄肼、甲基苄肼、普罗苄肼、普罗卡巴兴、异丙胺酰苄肼,属于烷基化类抗肿瘤药物.临床上主要用于治疗何杰金氏病,对于其他恶性肿瘤如恶性淋巴瘤肺癌多发性骨髓瘤等也有一定的疗效.丙卡巴肼属于前药,自身没有抗肿瘤作用,在体内经红细胞及肝微粒体酶的作用,氧化生成偶氮甲基苄肼,后者通过其N-甲基末端的转甲基作用,将甲基移转到鸟嘌呤的7位、腺嘌呤的1位或者t RNA的一些碱基上,烷化特定碱基,从而抑制DNA、RNA及蛋白质的合成,干扰肿瘤细胞的增殖.该药作用没有细胞周期特异性,但普遍认为丙卡巴肼主要阻碍S期细胞进入G2期[1-2].1982年该药的盐酸盐以口服胶囊剂型被FDA批准在美国上市,用于恶性淋巴瘤的治疗.
针对丙卡巴肼现有合成工艺[3]存在的缺点,即步骤较多,反应过程需要高温溴化取代和强酸氧化回流等条件,对设备损耗大;而且硫酸甲酯的使用对环境安全又造成较大威胁.本课题组曾报道了一个改进的工艺路线[4],但该工艺路线步骤仍然比较多(5步),第3步苄溴的制备也存在不安全因素.因此,本实验拟对该工艺路线进一步优化,路线见图1.
具体地,本实验采用对甲基苯甲酸为起始原料,经氯化亚砜回流得酰氯产物,再与异丙胺反应得N-异丙基对甲苯甲酰胺,化合物经硝酸铈铵(CAN)氧化生成(4-N-异丙氨基甲酰基)苯甲醛,产物与硫酸甲肼缩合得中间体后以氰基硼氢化钠还原得最终产物丙卡巴肼.总收率44.7%,单步收率71%~85%.
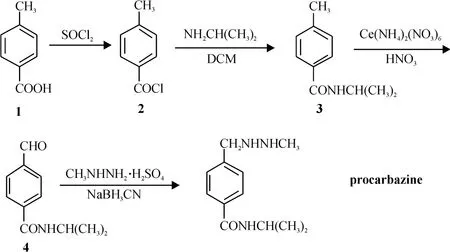
图1 丙卡巴肼的合成工艺再优化Figure 1 Re-optimization of the synthetic process for procarbazine
1 仪器和试剂
核磁共振谱用Bruker AV-300型核磁共振仪(Brucker Biospin公司,瑞士)测定(氘代氯仿,氘代甲醇为溶剂,TMS为内标).ESI-MS谱在Finnigan LCQ Advantage MAX质谱仪上测定.柱色谱用硅胶购自青岛海洋化工厂;硅胶GF-254薄层预制板购自烟台化学工业研究所;对甲基苯甲酸,硝酸铈铵(CAN)均购自上海晶纯试剂有限公司;二氯甲烷(DCM)经P2O5回流干燥,蒸馏备用;其他市售国产分析纯试剂都经过干燥纯化处理.
2 合成方法
对甲基苯甲酰氯(2):称取对甲苯甲酸136 mg(1.0 mmol)于10 mL的圆底烧瓶中,加入过量的二氯亚砜约3.0 mL,于80℃油浴加热回流3 h,减压蒸馏除去过量的二氯亚砜,得酰氯化合物,不经纯化,直接使用.
N-异丙基对甲苯甲酰胺(3):将酰氯化合物(2)重新溶于2.0 mL重蒸二氯甲烷,得A溶液;另取异丙胺0.5 mL(5.87 mmol)溶于1.0 mL干燥的二氯甲烷中,并将其缓慢地滴加到A溶液中,该过程温度控制在30℃下.加料完毕逐渐升至室温下搅拌反应3 h,之后升温至40℃,继续反应30 min,停止加热将反应液倾入25 mL饱和氯化钠水溶液中,用30 mL二氯甲烷萃取4次,合并有机相,无水硫酸钠干燥,旋蒸除去溶剂得白色固体151.2 mg,TLC检测为纯化合物,收率85.4%.1H NMR(300 MHz,CDCl3)δ:7.67(d,J=9.0 Hz,2H),7.20(d,J=9.0 Hz,2H),4.19~4.34(m,1H),2.37(s,3H),1.25(d,J=6.0 Hz,3H),1.23(d,J=6.0 Hz,3H);13C NMR(75 MHz,CDCl3)δ:166.7,141.6,132.2,129.1(2),126.9(2),41.8,22.9,21.4(2);ESI-MS(m/z):178.3[M+H]+,200.3[M+Na]+.数据与文献[4]同.
4-甲酰基-N-异丙基苯甲酰胺(4):取5 mL重蒸馏水与6 mL浓硝酸混合均匀,制成约3.5 mol/L的浓硝酸备用.称取N-异丙基对甲苯甲酰胺177 mg(1.0 mmol)于耐压管中,加入2 mL 3.5 mol/L硝酸溶液,超声震荡溶解.取硝酸铈铵4.4 g(4.0 mmol)于西林瓶中,加入8.0 mL 3.5 mol/L硝酸溶液,搅拌5 min,体系为CAN的硝酸混悬液.取搅拌均匀的CAN混悬液逐滴滴加到耐压管中,加料完毕,置于100℃油浴锅中反应24 h.反应结束,将反应液冷却,随后加入100 mL的NaCl饱和水溶液,用DCM萃取(3×100 mL),合并有机相,加入无水硫酸钠干燥,过滤,旋干有机溶剂得微黄色固体粉末,用硅胶柱色谱纯化(乙酸乙酯-石油醚,体积比1∶5)得白色固体136.2 mg,收率71.2%.1H NMR(300 MHz,CD3OD)δ:10.03(s,1H),7.95(d,J=9.0 Hz,2H),7.58(d,J=9.0 Hz,2H),4.10-4.30(m,1H),1.26(d,J=6.0 Hz,3H),1.24(d,J=6.0 Hz,3H);13C NMR(75 MHz,CD3OD)δ:193.5,168.4,130.6,129.0,128.1(2),127.2(2),43.4,22.5(2);ESI-MS(m/z):192.3[M+H]+,224.5[M+Na]+.
丙卡巴肼(procarbazine):称取化合物(4)191 mg(1.0 mmol)和甲基肼硫酸盐505 mg(3.5 mmol)于250 mL反应瓶中,加入无水乙醇20.0 mL,搅拌溶解,搅拌20 min后,加入三乙胺1 mL,将反应装置置于60℃油浴锅中反应6 h,反应结束,旋干溶剂,加入10 mL的DMF将其重新溶解,在0℃的低温恒温器中缓慢加入氰基硼氢化钠126 mg(2.0 mmol),加料完毕,反应体系逐渐升至室温,反应过夜.反应结束后,加水猝灭反应.加入200 mL的NaCl饱和水溶液,用200 mL的乙酸乙酯萃取5次,合并有机相,加入无水硫酸钠干燥,过滤,合并有机相,旋干得黄色油状液体.硅胶柱色谱分离(乙酸乙酯-石油醚,体积比1∶3),得丙卡巴肼161.9 mg,收率74%.1HNMR(300 MHz,CD3OD)δ:7.81(d,J=7.2 Hz,2H),7.24(d,J=7.2 Hz,2H),3.88-3.91(m,1H),3.93(s,2H),2.47(s,3H),1.25(d,J=6.0 Hz,3H),1.23(d,J=6.0 Hz,3H);13C NMR(75 MHz,CD3OD)δ:167.2,136.3,135.3,128.6(2),127.7(2),51.5,42.4,34.1,21.1(2).数据与文献[4]同.
3 结果与讨论
由于环境友好、生产成本低的对二甲苯空气氧化法[5]的投入使用,对甲基苯甲酸成为廉价的有机化工原料,因此,选择对甲基苯甲酸作为合成丙卡巴肼的起始原料是较佳的选择.通过与过量二氯亚砜制备酰化物2[6-7],是一条清洁的反应路线,过量的二氯亚砜可以回收,循环利用;过程中产生的盐酸气和SO2可以通过气体吸收塔吸收,避免污染环境,同时也可变废为宝;通过使用过量的异丙胺与2缩合,高收率地得到酰胺化合物3.过量异丙胺在实际生产过程中,可以回收,循环利用.

表1 氧化剂、溶剂及其他反应条件的选择Table 1 Selection of oxidant,solvent and other conditions
由化合物3合成化合物4,即苄基氧化成醛,是本工艺路线的关键步骤.文献[8-16]已报道了多个实验方法,包括硝酸铈铵(CAN)、PCC、SeO2、CrO3-Ac2O都曾被成功应用于苄基的氧化.本实验发现,对于4-甲基-N-异丙基苯甲酰胺底物体系,CAN是氧化苄基的最佳氧化剂;另外,反应介质的选择也相当重要(表1).实验结果揭示,以50%质量浓度的乙酸溶液作为反应溶剂,能够发生氧化反应,但难以使反应完全,产率30%~60%,且随着温度升高,副产物越来越多;以3.5 mmol/L的硝酸溶液作溶剂,可取得最佳的氧化效果,在100℃加热24 h后,原料已基本反应完全,反应收率可达72%(表1,No.10).最后采用还原氨化的方法将化合物4转化为丙卡巴肼,该反应是一个反应条件较为温和的过程,收率74%.
综上所述,本实验以对甲基苯甲酸和甲基肼硫酸盐为原料以4步反应合成丙卡巴肼,总收率44.7%(以对甲苯甲酸计).该工艺简化了合成方法,单元反应收率较高、条件温和,可在一定程度上降低能耗,节约资源.
[1]SINLIA B K.Metabolic activation of procarbazine.Evidence for carbon-centered free-radical intermediates[J].Biochem Pharmacol,1984,33(17):2777-2781.
[2]HORSTMAN M G,MEADOWS G G,YOST G S.Separate mechanisms for procarbazine spermato toxicity and anticancer activity[J].Cancer Res,1987,47(6):1547-1550.
[3]谭卫宁,刘智凌.水合肼的衍生物及其应用[J].湖南化工,1996,26(4):5-8.
[4]刘志军,陈河如.抗癌药物丙卡巴肼的合成工艺优化研究[J].中国药物化学杂志,2012,22(6):499-502.
[5]宋 磊,李 静,陈勇强,等.对二甲苯液相空气氧化的研究[J].聚酯工业,2007,20(2):22-24.
[6]BABU V V S,VASANTHAKUMAR G R,TANTRY S J.N-Silylation of amines and amino acid esters under neutral conditions employing TMS-Cl in the presence of zinc dust[J].Tetrahedron Letters,2005,46(38):4099-4102.
[7]PRAJAPATI A K,MODI V P.Synthesis and biological activity of N-[5-(4-methylphenyl)diazenyl-4-phenyl-1,3-thiazol-2-yl]benzamide derivatives[J].Quimica Nova,2001,34(5):771-774.
[8]SUGIYAMA Y,KURATA Y,KUNDA Y,et al.A fluorous Mukaiyama coupling reagent for a concise condensation reaction:utility of medium-fluorous strategy[J].Tetrahedron,2012,68(20):3885-3892.
[9]MATSUGI M,HASEGAWA M,SADACHIKA D,et al.Preparation and condensation reactions of a new light-fluorous Mukaiyama reagent:reliable purification with fluorous solid phase extraction for esters and amides[J].Tetrahedron Letts,2007,48(38):4147-4150
[10]RAHMAN O,KIHLBERG T,LANGSTROM B.Aryl riflates and[11C]/[13C]carbon monoxide in the synthesis of 11C-13C-amides[J].J Org Chem,2003,68(9):3558-3562.
[11]AlTALIB M,JOCHIMS J C.The reaction of acylium salts with carbodiimides[J].Chemische Berichte,1985,118(4):1304-1314.
[12]LEI M,MA L,HU LI.A convenient one-pot synthesis of formamide derivatives using thiamine hydrochloride as a novel catalyst[J].Tetrahedron Letters,2010,51(32):4186-4188.
[13]DUST L A,GILL E W.Nitrate Esters as Intermediates in the Oxidation of Toluenes by Ammonium Cerium(Ⅳ)Nitrate[J].J Chem Soc(C),1970,1630-1670.
[14]ZHAO H.The synthesis and structures of deuterium-labeled 5-substituted 1H-tetrazoles[J].J Label Compd Radiopharm,2008,51:293-296.
[15]HOSSEINZADEH R,TAJBAKHSH M,VAHEDI H.Selective oxidation of methylarenes with pyridinium chlorochromate[J].Synlett,2005,18:2769-2770.
[16]姜 华,陈蓉蓉,蒲含林.长春西汀的半合成工艺[J].暨南大学学报:自然科学版,2012,33(1):65-68.
[责任编辑:刘蔚绥]
Re-optimization of the synthetic process for antineoplastic drug procarbazine
PAN Long1,WANG Yawei1,LIU Xiaotian1,CHEN Heru1,2
(1.Institute of Traditional Chinese Medicine&Natural Products,College of Pharmacy;2.Guangdong Province Key Laboratory of Pharmacodynamic Constituents of TCM and New Drugs Research,Jinan University,Guangzhou 510632,China)
Procarbazine has been prepared through a four-steps process.Firstly,4-methylbenzoyl chloride was synthesized by reaction of 4-methylbenzoic acid with dimethyl sulfoxide.The direct reaction of this chloride with isopropylamine form N-isopropyl-4-methylbenzamide.The amide was then oxidized to 4-formyl-N-isopropylbenzamide by using ammonium ceric nitrate as oxidant.Afterwards,this intermediate condensed with methylhydrazine sulfate and then reduced by sodium cyanoborohydride under basic condition to offer the target compound.The overall yield was 44.7%.The yield of each unit reaction was 71% to 86%.All the intermediates and target product were identified by1H NMR,13C NMR and mass spectroscopy(MS).
procarbazine;synthetic process;drug synthesis;anticancer
O629.7
A
1000-9965(2015)01-0007-04
10.11778/j.jdxb.2015.01.002
2014-08-02
国家自然科学基金项目(81172982)
潘 龙(1988-),男,研究方向:新药设计与合成
陈河如(1967-),男,教授,博士生导师,研究方向:药物化学,Tel:020-38375299,E-mail:thrchen@jnu.edu.
