一个中国G6PD缺乏症家系致病基因的突变分析
2015-07-31林美华陈业达欧阳平谭艺青赵小蕾覃继恒饶绍奇
林美华,陈业达,欧阳平,赵 翔,谭艺青,赵小蕾,覃继恒,饶绍奇
研究论文
一个中国G6PD缺乏症家系致病基因的突变分析
林美华#,陈业达#,欧阳平,赵 翔,谭艺青,赵小蕾,覃继恒,饶绍奇*
(广东医学院 医学系统生物学研究所与公共卫生学院, 广东 东莞 523808)
目的对一个葡萄糖-6-磷酸脱氢酶(G6PD)缺乏症家系成员G6PD基因13个外显子全面测序,识别致病的突变位点及其遗传模式。方法在G6PD缺乏症高发区广东惠州地区收集到一个G6PD缺乏症核心家系,包括先证者(儿子)、患病母亲和正常父亲。取家系成员的外周血样,并提取基因组DNA,用PCR和DNA测序法对G6PD基因全部外显子进行序列分析。结果先证者及其母亲在G6PD基因第2号外显子出现同一点突变(c.95A>G,p.His32Arg),该突变引起组氨酸被精氨酸替换(CAC>CGC)。然而先证者父亲在G6PD基因的13个外显子均未出现突变。3种生物信息学软件均预测该突变对蛋白质功能具有较强的危害性。结论该家系中c.95A>G呈X连锁显性遗传,是G6PD缺乏症的致病突变位点之一。
葡萄糖-6-磷酸脱氢酶;基因突变;外显子测序
葡萄糖-6-磷酸脱氢酶(glucose-6-phosphate dehydrogenase, G6PD)缺乏症为一种常见的伴性不完全显性遗传病,常见于中国西南和华南地区,其受累人数多[1]。G6PD对细胞内自由基和H2O2的清除起着重要的作用[2],其基因突变导致的缺陷会引起红细胞膜的损害,导致相应的疾病。有研究报道G6PD缺陷症的患者中有多种突变基因型[3],然而,像这些关于G6PD基因突变位点的研究多数是基于群体的筛查,鲜有基于家系的遗传分析。本研究利用在广东惠州地区收集到一个G6PD缺乏症核心家系,通过对该家系成员的G6PD基因13个外显子全面测序,识别出致病的突变位点为c.95A>G,呈X连锁显性遗传。该结果将有助于华南地区G6PD缺乏症的基因诊断和产前咨询。
1 材料与方法
1.1 研究对象
本实验室研究人员在广东省惠州市人民医院收集一个家系共3人,儿子(先证者)及其母亲确诊为G6PD缺乏症,其父亲为正常个体。研究人员采集此家系人员的血液样本,同时调查了该家系成员的临床信息,采血和调查之前均获得家庭成员的知情同意。
1.2 主要试剂
DNA提取试剂盒(Tiangen公司);PCR试剂盒(TaKaRa公司);核酸电泳DNA marker(北京索莱宝科技有限公司)。
1.3 全血基因组DNA的提取
抽取G6PD缺乏症患者4 mL外周血,用DNA提取试剂盒提取全血基因组DNA,严格按试剂盒说明书操作。
1.4G6PD基因的扩增
9对上下游引物涵盖了X染色体上G6PD基因的13个外显子,其中用第3对引物扩增第3、4号外显子,用第8对引物扩增第10、11号外显子,用第9对引物扩增第12、13号外显子。G6PD基因的13个外显子中,最小外显子为第3号外显子,其长度为38 bp,最大的外显子为第13号外显子,长度为700 bp(图1)。PCR反应体系为:0.5 μL TaKaRa LA Taq、25 μL缓冲液、8 μL dNTPs、1 μL DNA、正反引物各1 μL,ddH2O补足体积至50 μL。PCR反应条件为94 ℃变性30 s,56 ℃退火30 s,72 ℃延伸2 min,扩增35个循环。PCR产物经1%琼脂糖凝胶电泳鉴定。
1.5 DNA测序和突变分析
用双脱氧链终止法技术对样品进行测序。采用英潍捷基公司3730XL测序仪对PCR产物进行测序,得到的序列与GenBank中标准参考序列进行比对,并使用软件BioEdit、ClustalX 2.1对序列进行分析。突变位点参考HGVS(http://www.hgvs.org/)标准命名。
1.6 突变危害性的预测
用PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)、Align GVGD(http://agvgd.iarc.fr/)和MutationTaster(http://www.mutationtaster.org/)软件预测突变对蛋白质功能的影响程度。PolyPhen-2是基于蛋白质三维结构的原理,Align GVGD基于氨基酸的生物学特性和蛋白质的多序列比对的原理,MutationTaster则是基于氨基酸序列同源性的原理来对突变进行预测。仅当两种或以上软件均预测同一突变对蛋白质的功能影响较大时,才认为该突变具有较强的危害性。
2 结果
2.1 家系临床资料分析
患者(先证者)食蚕豆后1 d发病,有头晕、发热、 倦怠无力、 厌食、 恶心和不定性的腹痛等早期症状,出现皮肤巩膜黄染、贫血以及酱油色尿,遂来医院就诊。血液检查提示:1)血红蛋白急剧下降;2)红细胞最低降至0.5×1012/L以下;3)网织红细胞明显增高>0.20;4)外周血涂片可见有核红细胞增多;5)白细胞数升高。生化检查提示:葡萄糖-6-磷酸脱氢酶活性减低,临床诊断为G6PD缺乏症。此家系中,先证者母亲及先证者先后被诊断出具有典型的临床症状,表现为皮肤巩膜黄染、贫血以及酱油色尿,并且实验室检查完全符合诊断标准。先证者父亲未曾出现G6PD缺乏症的临床症状,其血液生化学检查,显示是正常的个体。

Black blocks represent exons and lines represent introns in this picture图1 G6PD基因的结构Fig 1 Structure of a G6PD gene
2.2G6PD基因突变分析
先证者及其母亲在第2号外显子出现同一点突变(c.95A>G,p.His32Arg),该突变引起组氨酸被精氨酸替换(CAC>CGC);而父亲序列未见突变(图2)。
2.3 生物信息学分析
p.His32Arg突变很可能对蛋白质正常功能产生有害作用(表1)。
3 讨论
G6PD缺乏症在地理位置上分布广泛,罹患者遍及全球,估计世界各地约有4亿人受累,其危害较大[5]。G6PD是磷酸戊糖旁路途径的第一个限速酶,能催化6-磷酸葡萄糖生成还原型NADPH[6]。由于成熟的红细胞缺乏线粒体及细胞核,只能通过磷酸戊糖旁路生成NADPH。NADPH能够使氧化型的谷胱甘肽转化为还原型的谷胱甘肽。还原型谷胱甘肽对于红细胞内H2O2和自由基的清除起着重要的作用[7],当体内H2O2和自由基等氧化性的物质产生过多或清除障碍时,可对细胞膜蛋白质、脂质和细胞内的酶产生氧化损伤,这些损伤可导致组织细胞结构和功能的异常。
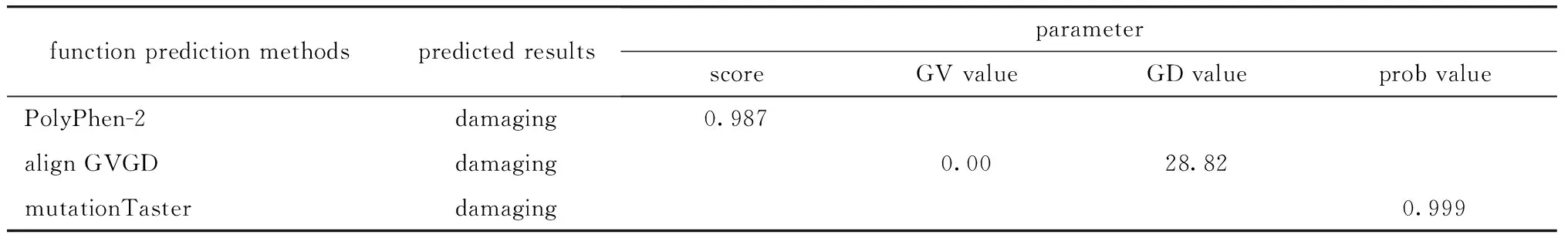
表1 生物信息学预测突变对蛋白质功能的影响Table 1 Predicting the effects of a mutation on protein function by bioinformatics
The greater the score and prob value ranging from 0 to 1, the greater the damaging; If(GV=0) and (GD> 0).the position of interest is invariant (100% conservation) so any mutation at the position is predicted as damaging[4].
G6PD基因中第2号外显子中的碱基A突变为G,可导致氨基酸的错义突变,造成G6PD上的第32位氨基酸发生改变,由组氨酸变成精氨酸,进而导致蛋白的结构功能发生变化,最终影响G6PD的酶活性。有研究报道G6PD缺陷症患者的G6PD基因上的突变位点为c.95A>G[8-9],本研究也得出了相同的结果。为了进一步提供更强的证据阐明该位点的突变对蛋白质功能的影响,本研究使用PolyPhen-2、Align GVGD和MutationTaster对其进行生物信息学分析,预测该位点的突变是否对蛋白功能产生有害作用。3种方法预测结果都显示了该位点的突变对蛋白的功能将产生有害作用,因此可认为该突变具有较强的危害性。
本次研究检测到先证者及其母亲G6PD基因第2号外显子的一个错义突变(c.95A>G),该突变位点在以往研究中已有报道[10-11],在dbSNP数据库编号为rs137852340。此家系中,先证者母亲为杂合子,携带杂合型G6PD突变型等位基因c.95A>G,先证者为半合子,X染色体上携带G6PD突变型等位基因来自于其母亲,其父亲正常,这符合G6PD缺乏症伴性显性遗传的特点。但由于研究的数据量偏小,不能估计其外显率,也无法对其遗传机制进行严格的统计分析。本次研究通过对一家系中G6PD缺陷患者进行基因突变的鉴定及其临床表现的观察,推测了G6PD基因的结构和功能之间可能存在的关系,基因突变与疾病发病的具体遗传机制需要在更大的家系中进一步验证。
志谢:本研究承蒙医院各医生的支持与协助!
[1] 徐芸,罗建明. 我国G6PD缺乏症基因突变的研究现状[J]. 中国小儿血液与肿瘤杂志, 2009, 03: 143-145.
[2] Wang YP, Zhou LS, Zhao YZ,etal. Regulation of G6PD acetylation by KAT9/SIRT2 modulates NADPH homeostasis and cell survival during oxidative stress[J]. Eur Mol Biol Organ, 2014, 33: 1304-1320.
[3] 于国龙,蒋玮莹,杜传书,等. 广东客家人G6PD基因突变型研究[J]. 中华医学遗传学杂志, 2004, 05: 32-35.
[4] Mathe E, Olivier M, Kato S,etal. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods[J]. Nucleic Acids Res, 2006, 34: 1317-1325.
[5] Nkhoma ET, Poole C, Vannappagari V,etal. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: A systematic review and meta-analysis[J]. Blood Cells Mol Dis, 2009, 42: 267-278.
[6] Kotaka M, Gover S, Vandeputte-Rutten L,etal. Structural studies of glucose-6-phosphate and NADP[J]. Acta Crystallographica Section D Biol Crystallography, 2005, 61: 495-504.
[7] Zou CG, Agar NS, Jone GL. Oxidative insult in sheep red blood cells induced by T-butyl hydroperoxide: the roles of glutathione and glutathione peroxidase[J]. Free Radic Res, 2001, 34: 45-56.
[8] 王也飞,夏文权,倪培华,等. G6PD缺陷症基因型分析:一种新的错义突变[J]. 上海交通大学学报:医学版, 2010, 06: 698-702.
[9] 蔡望伟,周玉英,周代锋,等. 海南汉族、黎族人葡萄糖-6-磷酸脱氢酶缺乏症的基因突变型分析及一种新的G6PD基因突变型的鉴定[J]. 中华医学遗传学杂志, 2001, 18: 105-109.
[10] Chao LT, Du CS, Louie E,etal. A to G substitution identified in exon 2 of the G6PD gene among G6PD deficient Chinese[J]. Nucleic Acids Res, 1991, 19: 6056-6056.
[11] Beutler E, Vulliamy TJ. Hematologically important mutations: glucose-6-phosphate dehydrogenase[J]. Blood Cells Mol Dis, 2002, 28: 93-103.
Mutation analysis of the pathogenic gene in a Chinese family with G6PD deficiency
LIN Mei-hua#, CHEN Ye-da#, OUYANG Ping, ZHAO Xiang, TAN Yi-qing,ZHAO Xiao-lei, QIN Ji-heng, RAO Shao-qi*
(Institute for Medical Systems Biology and School of Public Health, Guangdong Medical College, Dongguan 523808, China)
Objective In order to recognize the mutation site as well as its inheritance model. Methods We collected the family trio with G6PD deficiency in Huizhou of Guangdong Province, a region of high prevalence of G6PD deficiency. The family trio includes the proband, his affected mother and healthy father.Their blood samples were collected and genomic DNA was extracted. Sequence analysis was performed in thirteen exons by PCR and DNA sequencing. Results The results indicated that the proband and his mother shared an identical mutation (c.95A>G, p.His32Arg) in exon 2 ofG6PDgene. This mutation caused histidine to be replaced with arginine (CAC>CGC). However, no single mutation was found in his father. Three bioinformatics tools predicted that this mutation was detrimental to the function(s) of its associated protein. Conclusions The mutation, c.95A>G, following an X-linked dominant inheritance model, is a causal site for G6PD deficiency.
glucose-6-phosphate dehydrogenase; gene mutation; exon sequencing
2015-01-28
2015-04-27
国家自然科学基金(31071166,81373085);广东省科技计划攻关项目(2009A030301004);东莞市科技重点项目(2011108101015)
*通信作者(corresponding author):raoshaoq@gdmc.edu.cn
#对本文有相同贡献
1001-6325(2015)08-1011-04
R394.3
A
