儿童Gitelman 综合征的SLC12A3 基因复杂杂合突变
2015-05-07高春林马上茹夏正坤高远赋樊忠民莫桂玲
高春林,马上茹,夏正坤,高远赋,樊忠民,徐 敏,魏 伟,周 昱,莫桂玲
0 引 言
儿童Gitelman 综合征(gitelman syndrome,GS)是由定位于16 号染色体长臂(16q13)编码肾远曲小管钠-氯协同转运蛋白(Na-Cl cotransporter,NCCT)的SLC12A3 基因突变所致的综合征,属常染色体隐性遗传性疾病。其临床表现为低血钾、低血镁、碱中毒及继发性高肾素高醛固酮血症,患儿可出现乏力、便秘、头晕、肌肉痛、生长延迟等低血钾症状[1],肾活检可表现为肾小球球旁器增生或正常。因低血钾的持续存在,在合并呕吐、腹泻等突发情况时,可致Q-T 间期延长,严重时可出现心律失常、横纹肌溶解、晕厥、猝死等危险情况,具有潜在的危险性。目前发病率为1/40 000[2],据来自印度的资料,该病居致儿童死亡病因的第4 位[3-4],是少见但可能严重威胁儿童健康的疾病之一。本文对2 例儿童GS 进行了研究,现报道如下。
1 资料与方法
1.1 研究对象 全面收集2013 年1 月至2014 年5月在南京军区总医院儿科住院的2 例低血钾患儿临床资料。其中男性1 例,13 岁;女性1 例,8 岁;均为汉族,生长发育均在正常范围,无特异症状。男性患儿因需行扁桃体切除术,术前常规检查发现低血钾(2.8 mmol/L);女性患儿因患支气管肺炎门诊检查时发现低血钾(2.4 mmol/L)。为明确诊断对患儿行DNA 测定,本研究通过医院伦理委员会批准,经患儿家属同意并签署知情同意书。
1.2 DNA 测序(一代测序法)
1.2.1 提取基因组DNA 抽取受检者的外周血3 mL,采 用 QIAamp 外 周 血 DNA 提 取 试 剂 盒(QIAamp,德国),按其操作说明书提取基因组DNA。
1.2.2 获取SLC12A3基因序列 从美国国家生物技术信息中心(the national center for biotechnology information,NCBI)数据库获取人类SLC12A3 基因DNA 序列(网址http://www.ncbi.nlm.nih.gov/gene/?term=SLC12A3)。
1.2.3 聚合酶链反应(polymerase chain reaction,
PCR) 利用Primer Premier 5 软件(网址http://www.bbioo.com/download/58-166-1.html)自行设计能够扩增SLC12A 基因外显子编码区及剪切位点的引物,并由英潍捷基(上海)贸易有限公司合成。引物序列见表1。
PCR 条件为:95 ℃预变性5 min;94 ℃变性30 s;65 ℃退火1 min;72 ℃延伸1 min,共35 个循环,最后72 ℃延伸7 min,25 ℃保存。PCR 产物测序:采用Big Dye Terminatorv3.1 循环测序试剂盒(Applied Biosystems,美国),按其操作说明书进行测序。
1.2.4 测序结果分析 测序结果用Mutation Surveyor V4.0.5(demo)软件(美国Softgenetics 公司)分析,所得序列与NCBI 数据库(网址http://www.ncbi.nlm.nih.gov/gene/?term=SLC12A3)中的SLC12A3 基因序列NG_009386.1 进行比对。碱基变异参考人类基因组变异研究学会(human genome variation society,HGVS)制定的序列变异命名(www.genomic.unimelb.edu.au/mdi/mutnomen/)的命名法则,以GenBank cDNA 序列NM_000339.2 的翻译起始密码子ATG 中的A 为+1 位核苷酸进行命名。
1.2.5 多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)检测 MLPA 检测试剂盒“SALSA MLPA P136 Gitelman syndrome probemix”购自荷兰MRC-Holland,按照试剂盒说明书进行检测。
1.2.6 MLPA 检测结果分析 使用Coffalyser v9.4软件进行结果分析,评定标准为缺失外显子的信号全部消失,Ratio 为0;重复区表现出信号成倍增加,Ratio 约为2.00;杂合缺失相应的外显子缺失信号将降低35%~55%,Ratio 为0.45 ~0.65;而杂合重复突变时相应的信号将升高30%~55%,Ratio 为1.30 ~1.55。
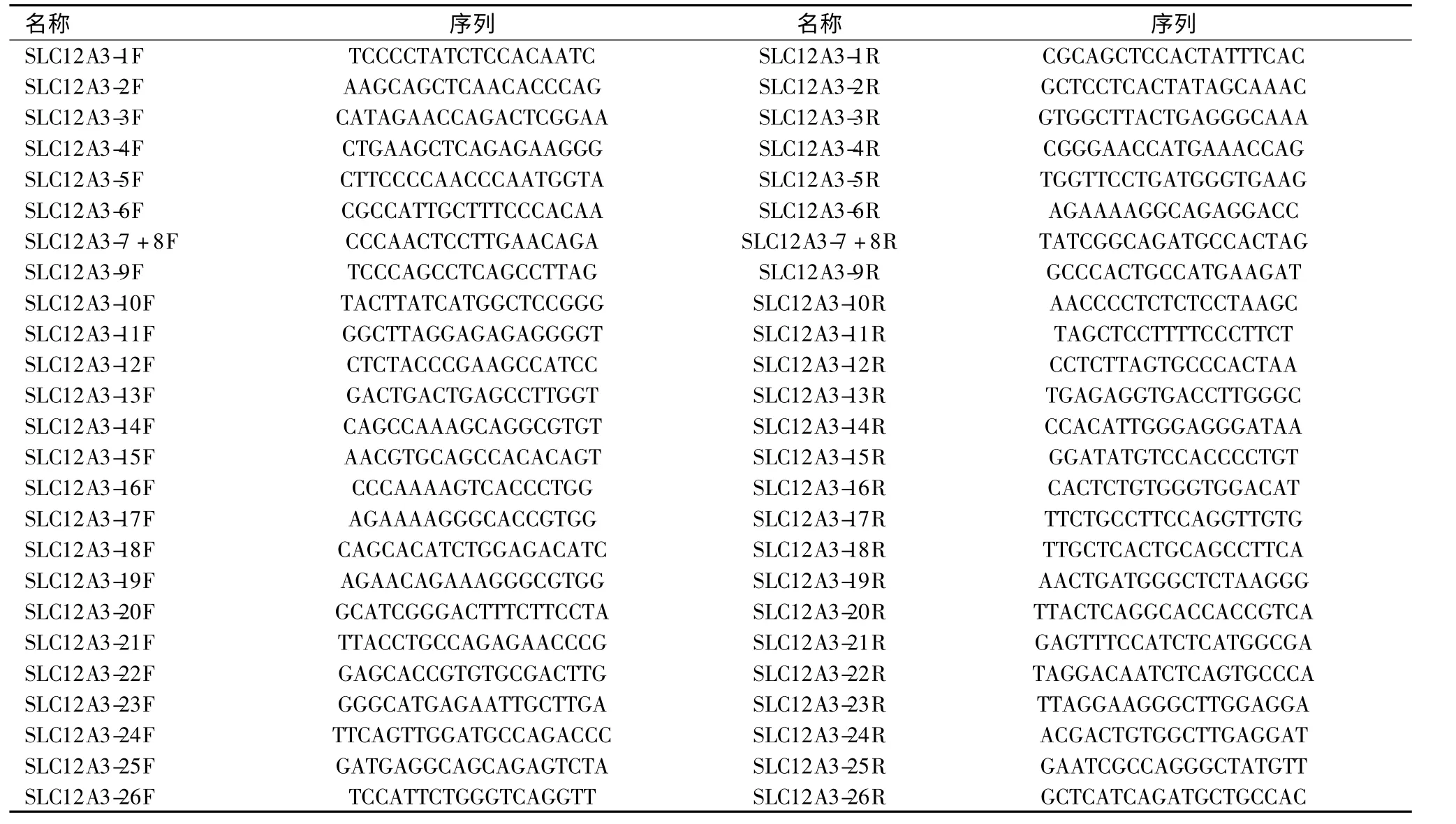
表1 SLC12A3 基因各外显子的引物序列Table 1 The Primer of SLC12A3 used in the study
2 结 果
2.1 男性患儿的SLC12A3 基因检测结果 应用一代测序方法检测到SLC12A3 基因的1 个杂合突变:c.1964G >A,p.(Arg655His);1964 位碱基G 突变为A,同时检测到多个并不致病的多态性位点(rs10927887,纯合;rs200268763,纯合等),应用MLPA方法检测到SLC12A3 基因8 号外显子的杂合缺失突变。SLC12A3 基因突变引起的GS 通常以常染色体隐性的方式遗传;从基因检测结果看来,受检者携带2 个杂合致病突变,符合BS/GS 患者的基因特征。经分析,c.1964G >A,p.(Arg655His)位点为已知突变,而8 号外显子缺失突变为新发现的突变,同时具有两者突变可致发病。见图1。
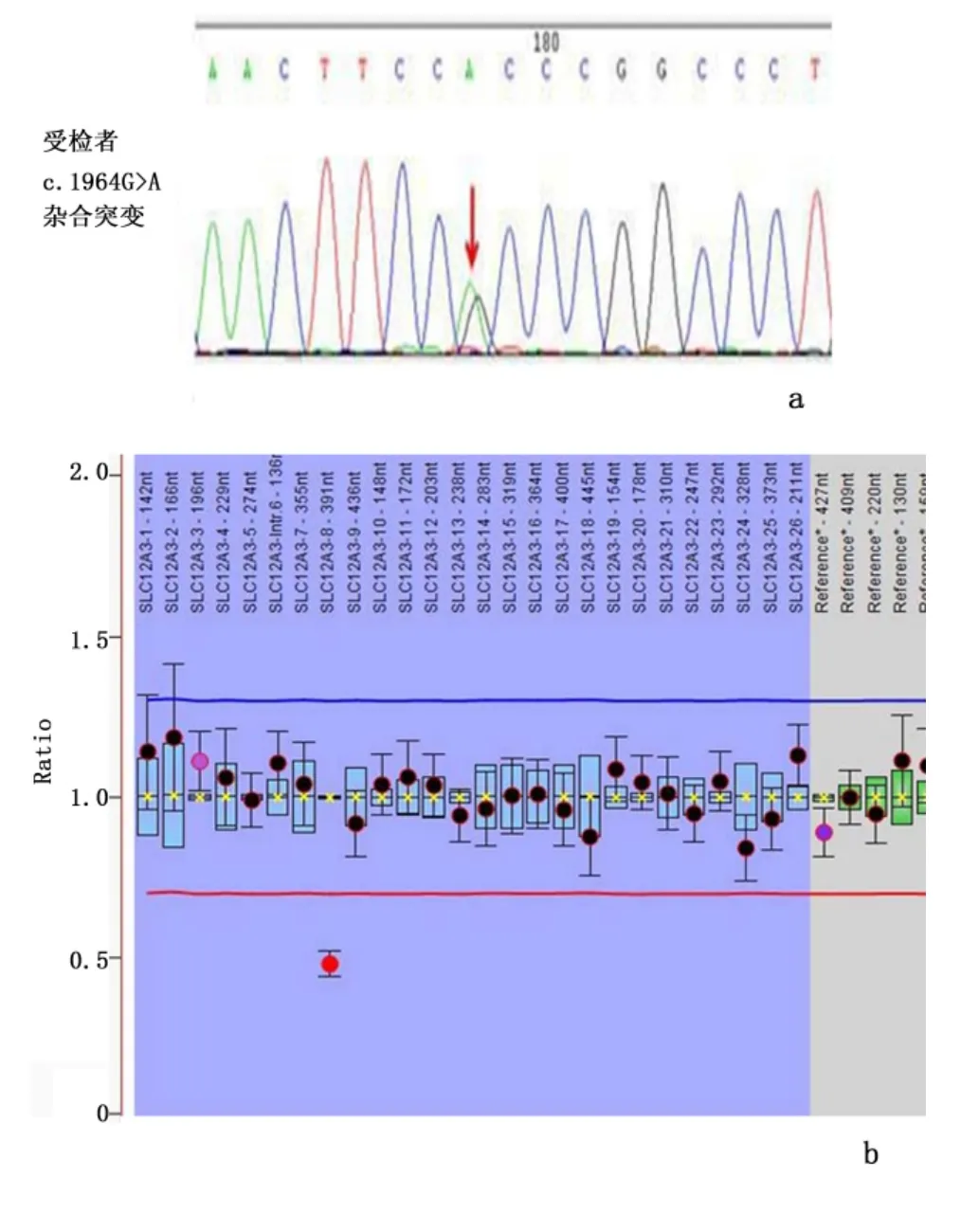
图1 男性患儿的SLC12A3 基因测序及SLC12A3 基因MLPA 结果Figure 1 SLC12A3 sequenced and MLPA results of the boy
2.2 女性患儿的SLC12A3 基因检测结果 应用一代测序方法检测到SLC12A3 基因的2 个杂合突变:c.2543A >T,p.(Asp848Val)和c.976delG,p.(Val326fs)突变;2543 位碱基G 突变为A,导致848 位门冬氨酸突变为缬氨酸,976 位碱基G 缺失,使氨基酸326 位缬氨酸后发生移码突变。其中c.2543A >T 突变未见文献报道,应用MLPA 方法未检测到SLC12A3 基因外显子的缺失突变。SLC12A3 基因突变引起的GS 通常以常染色体隐性的方式遗传;从基因检测结果看来,受检者携带2 个杂合致病突变,符合BS/GS 患者的基因特征。见图2。
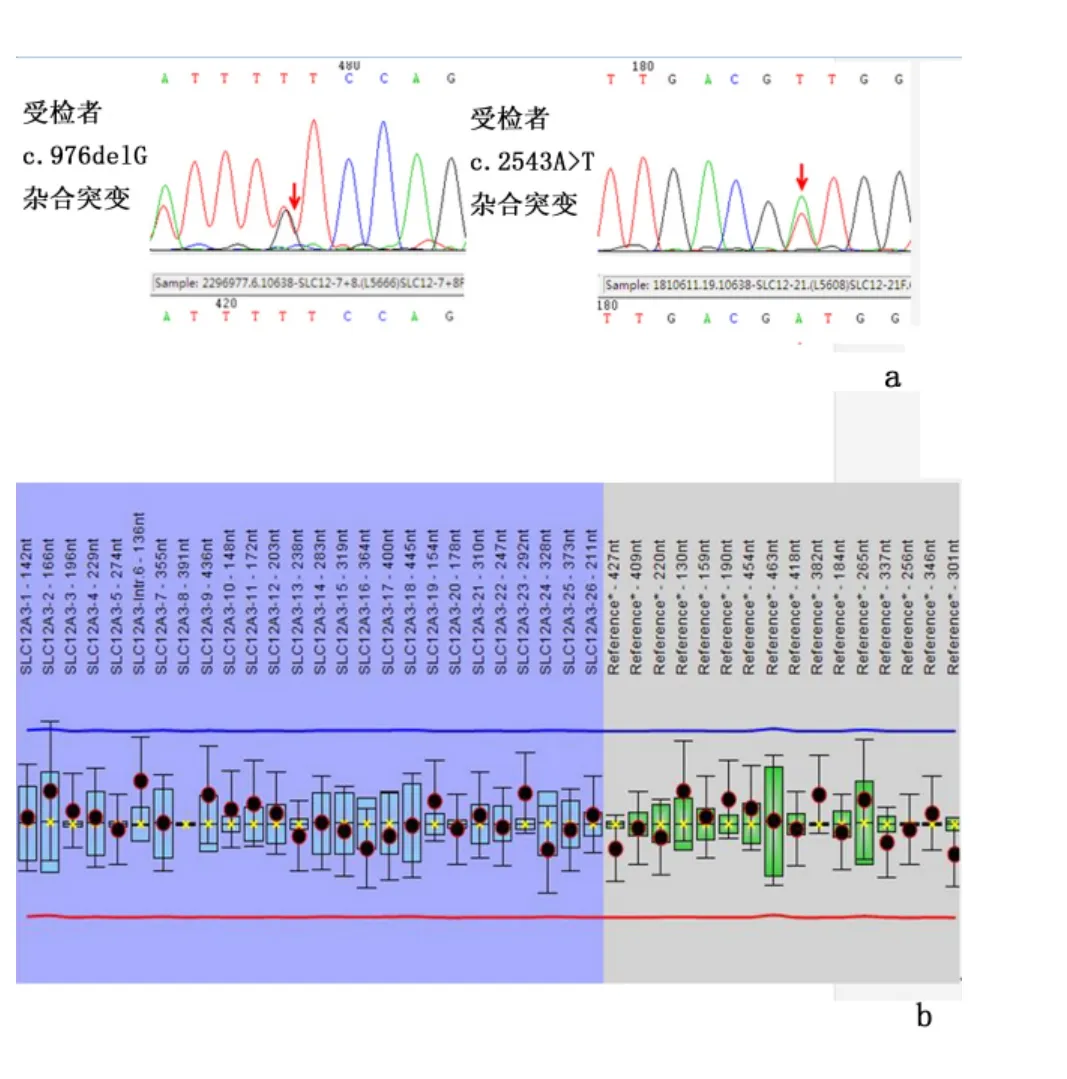
图2 女性患儿的SLC12A3 基因测序及SLC12A3 基因MLPA 结果Figure 2 SLC12A3 sequenced and MLPA results of the girl
3 讨 论
自1966 年Gitelman 首次报道3 例22 ~47 岁的女性患者并以GS 命名以来[5],世界各地每年均有数量不等的病例被诊断,尽管如此,儿童GS 的报道仍少见,儿科医师对此病认识不足,因而容易出现漏诊和误诊。
3.1 GS 与SLC12A3 基因突变 已知SLC12A3 基因定位于16q13,共含有26 个外显子,1030 个氨基酸,其二级结构包括跨膜区、细胞内氨基端和羧基端、细胞外疏水的环状区,其编码的蛋白质即为负责钠-氯共转运的载体蛋白NCCT,定位于远曲小管,该基因的异常突变即可致病。迄今为止,已发现超过250 种该基因的突变,某些突变可能导致NCCT 结构和/或功能异常[6]。Balavoine 等[7]分析了15 例GS 患者,其中9 例为纯合子突变,6 例为复合杂合子突变,并且突变位点个数表现严重度呈正相关。但相关儿童GS 的报道甚少。本研究发现2 例患者为复合杂合突变,其中c.1964G >A,p.(Arg655His)和c.2543A >T,p.(Asp848Val)突变位点均位于NCCT的细胞内羧基端,使其从尿液重吸收钠氯的功能受损,976 位碱基G 缺失,使氨基酸序列326 位缬氨酸后发生移码突变,蛋白质翻译异常,此为已知突变,可使NCCT 蛋白结构异常。此外发现8 号外显子的缺失突变可能导致NCCT 异常,目前尚无法预测,但该外显子应具有不可或缺的功能,其缺失或误义突变可能使得蛋白质翻译过程发生移码或提前终止从而丧失相应的功能。
3.2 基因检测是确诊的手段 本研究对2 例患儿进行了基因检测发现,男性患儿存在SLC12A3 基因位点的杂合突变,c.1964G >A,p.(Arg655His),该位点为已知突变;女性患儿存在2 个位点的基因突变,其中c.2543A >T,p.(Asp848Val)的突变尚未见报道,并通过美国国立卫生中心的单核苷酸多态性数据库排除该位点变异为单核苷酸多态性。由于GS 为常染色体隐性遗传,仅有1 个位点突变无法解释患者发病,本研究进一步行大片段缺失或重复突变的MLPA 检测,结果发现男性患儿同时表现SLC12A3 基因8 号外显子的杂合缺失突变;至此,根据患儿基因测序结果,明确诊断为复杂杂合突变所致的GS。既往研究显示:①复杂杂合突变是导致的GS 的重要突变模式,如Vargas-Poussou 等[8]对448例GS 进行研究发现,70%患者存在2 个位点突变,对具有1 个点突变的患者进行MPLA 检测后发现,其中47%的患者存在单个或多个外显子的大片段缺失或重复突变(即复杂杂合突变),与本研究的结果一致;②SLC12A3 基因的突变包含多种模式,无义突变、误义突变、剪切位点突变及缺失,如突变位点位于启动子区或剪切位点异常,异常的终止密码子等使翻译或蛋白加工或者折叠过程异常,NCCT 蛋白的完全缺失;甚至影响其嵌入胞浆膜进而缺乏正常的功能[6,9]。③8 号外显子的缺失突变尚未见报道,既往研究发现8 号外显子存在误义突变或移码突变等,甚至剪接缺失,但无大片段缺失的报道。因此MPLA 结合一代基因测序技术使得GS 的诊断更为精确和直接,并可与BS 相鉴别。本研究通过一代测序技术联合MPLA 检测,便于临床及时发现少见的突变,为诊断疾病提供有力的工具,有助于指导临床医师制定合理有效的治疗方案;2 例患儿确诊后给予积极补钾补镁治疗,并定期复查,维持血钾在正常范围。
3.3 GS 与BS 的鉴别诊断 典型的GS 表现除低血钾、代谢性碱中毒、RAS 激活、血压正常或偏低外,低尿钙、低血镁为其特点。因SLC12A3 基因异常导致NCCT 无法正确的转运钠,髓袢升支粗段和集合管则通过Na+/K+、H+/K+和Na+/H+代偿吸收钠,泌H+、泌K+增加,使得GS 患者只表现轻微的失盐,但低血钾明显,低血镁的机制目前认为是由于远曲小管钠依赖的镁离子重吸收降低导致[10];髓袢升支粗段代偿吸收钠增加使得正电荷电位明显增加,进而促进继发性的钙重吸收,低尿钙出现,这也是与BS 相区别的重要特点。在临床实践中应注重系统的血液及尿液检查,以利排除诊断或确诊。但也有研究发现,低镁血症不是诊断GS 的必要条件,部分患者血镁可正常,但低镁似与症状轻重相关[11]。
BS 典型的肾病理表现为肾小球球旁细胞器肥大,男性患儿未看到肾小球球旁复合体;女性患儿可看到个别球旁器肥大,电镜下足细胞无特殊改变。可见GS 也可表现为球旁细胞器肥大,提示该表现不具有鉴别GS 与BS 的特殊诊断价值。因而在具备基因诊断条件的情况下可酌情暂缓肾活检术。
综上所述,儿童GS 的临床表现及遗传学表现差异较大,需要儿科医师提高认识,掌握典型的临床表现,并积极进行基因诊断和遗传咨询。
[1] Gaur A,Ambey R,Gaur BK.Gitelman's syndrome:Rare presentation with growth retardation[J].Indian J Nephrol,2014,24(1):60-62.
[2] Sampathkumar K,Muralidharan U,Kannan A,et al.Childhood bartter's syndrome:an Indian case series[J].Indian J Nephrol,2010,20(4):207-210.
[3] Mhasde DR,Gautam S,Zore RR,et al.Gitelman's syndrome[J].J Assoc Physicians India,2009,57(3):211-212.
[4] Sundar U,Lakkas Y,Asole D,et al.Gitelman's syndrome presenting as recurrent paralytic ileus due to chronic renal tubular K+wasting[J].J Assoc Physicians India,2010,58(5):322-324.
[5] Gitelman HJ,Graham JB,Welt LG.A new familial disorder characterized by hypokalemia and hypomagnesemia[J].Trans Assoc Am Physicians,1966,79(1):221-235.
[6] Glaudemans B,Yntema HG,San-Cristobal P,et al.Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome[J].Eur J Hum Genet,2012,20(3):263-270.
[7] Balavoine AS,Bataille P,Vanhille P,et al.Phenotype-genotype correlation and follow-up in adult patients with hypokalaemia of renal origin suggesting Gitelman syndrome[J].Eur J Endocrinol,2011,165(4):665-673.
[8] Vargas-Poussou R,Dahan K,Kahila D,et al.Spectrum of mutations in Gitelman syndrome[J].J Am Soc Nephrol,2011,22(4):693-703.
[9] Gamba G.Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters[J].Physiol Rev,2005,85(2):423-493.
[10] Singh PJ,Nash JL,Santella RN,et al.Gitelman's syndrome:report of a 19-year old woman with intractable hypomagnesemia and hypokalemia and a review ofthe syndrome[J].S D J Med,1999,52(10):377-380.
[11] Jiang L,Chen C,Yuan T,et al.Clinical severity of gitelman syndrome determined by serum magnesium[J].Am J Nephrol,2014,39(4):357-366.
