新型苯并咪唑衍生物的合成及其抗凝血活性*
2015-04-23蔡志强刘若灿
蔡志强,侯 旭,张 波,刘若灿
(沈阳工业大学石油化工学院,辽宁辽阳 111003)
达比加群酯是一种新型的凝血酶抑制剂,用于口服的前体药物,属非肽类凝血酶抑制剂。其母核中苯并咪唑所连的芳环取代的脒基造成了药物的溶解性差和口服生物利用度低等缺点[1-4],限制了其药效的充分发挥。为获得活性更强,溶解性和生物利用度更佳的苯并咪唑类药物,本文整合了雷扎沙班及其类似物的设计理念,采用苯甲脒基环合形成苯并异恶唑,并将其另外一端侧链用氨基酸取代的方法优化其结构,增大化合物透膜吸收从而改善其药物代谢,并考察其构效关系。
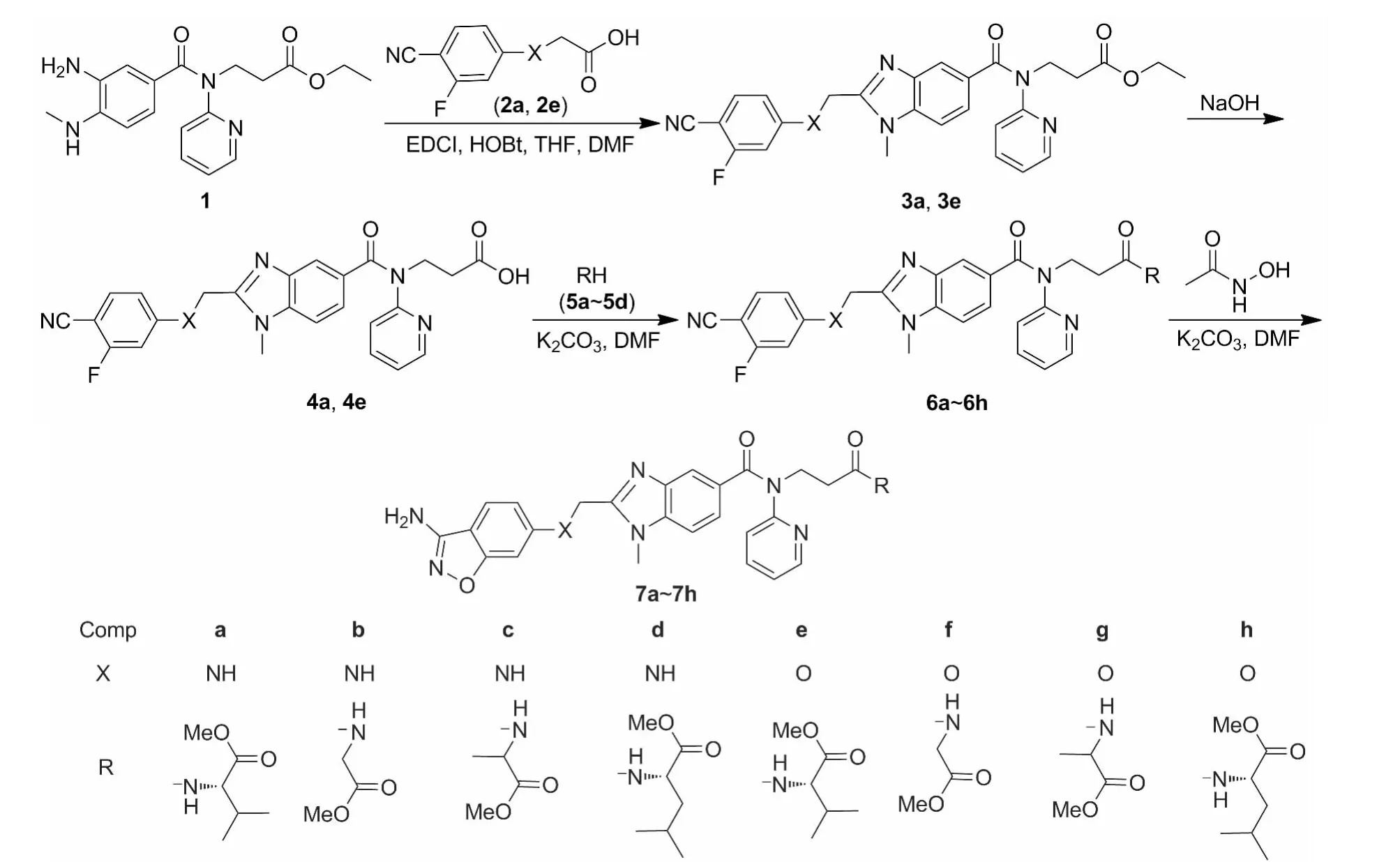
Scheme 1
在文献[5-10]方法的基础上,本文以 3-[(3-氨基-4-甲基氨基苯甲酰)吡啶-2-基氨基]丙酸乙酯(1)为原料,与4-氰基-3-氟苯取代基乙酸(2a,2e)经环化反应制得 3-【【2-{[(4-氰基-3-氟苯取代基)甲基]-1-甲基-1H-苯并咪唑-5-基}羰基】吡啶-2-基】氨基丙酸乙酯(3a和3e);3a和3e分别经水解和酰胺化反应制得 3-【【【2-{[(4-氰基-3-氟苯基)取代基]甲基}-1-甲基-1H-苯并咪唑-5-基】羰基】吡啶-2-基氨基】丙酰取代胺基(6a~6h);6与乙酰氧肟酸经环合反应合成了8个新型的苯并咪唑衍生物7a~7h(Scheme 1),其结构经1H NMR和HR-ESI-MS表征。并测定了7a~7h的抗凝血活性。
1 实验部分
1.1 仪器与试剂
Bruker ARX-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Agient1100型四级杆液质联用仪;Bruker micro-TOF型高分辨质谱仪。
1按文献[5]方法制备;其余所用试剂均为分析纯。
1.2 合成
(1)3a和3e的合成(以3a为例)
在反应瓶中依次加入2-[(4-氰基-3-氟苯基)氨基]乙酸(2a)1.94 g(10 mmol),1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDCI)1.9 g,1-羟基苯并三唑(HOBt)1.35 g,THF 35 mL和DMF 5 mL,冰水浴冷却,搅拌反应30 min。升至室温,缓慢滴加1 3.1 g(9 mmol)的 THF(15 mL)溶液,滴毕,反应6 h。除溶,加入二氯甲烷30 mL,用饱和盐水(3×15 mL)洗涤,无水硫酸镁干燥。过滤,滤液浓缩至干;剩余物中加入冰乙酸45 mL,回流反应2 h。减压浓缩至干,加入浓氨水15 mL,搅拌下于室温反应30 min。除溶,加入二氯甲烷25 mL,用饱和食盐水(3×15 mL)洗涤,无水硫酸镁干燥。过滤,滤液浓缩后经柱色谱[洗脱剂:A=V(CH2Cl2)∶V(MeOH)=20 ∶1]纯化得淡黄色固体3a 3.30 g,产率 72.8%;1H NMR δ:1.12(t,J=7.2 Hz,3H),2.69(t,J=7.2 Hz,2H),3.76(s,3H),3.98(q,J=7.2 Hz,2H),4.21(t,J=7.2 Hz,2H),4.63(d,J=4.4 Hz,2H),6.84 ~6.89(m,3H),7.11 ~7.15(m,2H),7.38 ~7.58(m,3H),7.67(d,J=8.4 Hz,2H),8.37(d,J=2.8 Hz,1H);ESI-MS m/z:501.3{[M+H]+}。
用类似方法合成土棕色固体3e。
(2)4a和4e的合成(以4a为例)
在反应瓶中加入3a 2.42 g(1.5 mmol)和乙醇30 mL,搅拌使其溶解;滴加氢氧化钠0.32 g(8 mmol)溶液5 mL,滴毕,反应3 h。蒸除1/3体积溶剂,用稀盐酸调至pH 6。过滤,滤饼干燥得白色固体 4a 1.82 g,产率 79.7%;1H NMR δ:2.69(t,J=7.2 Hz,2H),3.76(s,3H),3.98(q,J=7.2 Hz,2H),4.63(d,J=4.4 Hz,2H),6.84 ~6.89(m,3H),7.11 ~ 7.15(m,2H),7.38 ~7.58(m,3H),7.67(d,J=8.4 Hz,2H),8.37(d,J=2.8 Hz,1H),11.96(br,1H);ESIMS m/z:473.2{[M+H]+}。
用类似方法合成白色固体4e。
(3)6a~6h的合成(以6a为例)
在反应瓶中加入4a 4.72 g(0.01 mol),EDCI 1.9 g,HOBt 1.35 g,碳酸钾 1.38 g 和 DMF 25 mL,搅拌使其溶解;于冰水浴中反应1 h;升至室温,加入缬氨酸甲酯(5a)盐酸盐 1.68 g(10 mmol),反应6 h。除溶,加入二氯甲烷20 mL,用饱和盐水(3×10 mL)洗涤,无水硫酸镁干燥。过滤,滤液浓缩后经柱色谱(洗脱剂:A=25∶1)纯化得淡黄色固体 6a 3.60 g,产率61.5%;1H NMR δ:0.88(d,J=6.8 Hz,6H),2.32(m,1H),2.69(t,J=7.2 Hz,2H),3.70(s,3H),3.74(s,3H),3.98(q,J=7.2 Hz,2H),4.25(d,J=7.2 Hz,1H),4.63(d,J=4.4 Hz,2H),6.84 ~6.89(m,3H),7.11 ~ 7.15(m,2H),7.38 ~7.58(m,3H),7.67(d,J=8.4 Hz,2H),8.37(d,J=2.8 Hz,1H);ESI-MS m/z:586.1{[M+H]+}。
用类似方法合成灰白色固体6b~6h。
(4)3-【2-{[(3-氨基苯基异恶唑-6-基)取代基]甲基}-1-甲基-N-(吡啶-2-基)-1H-苯并咪唑-5-羰基】丙酰取代基(7a~7h)的合成(以7a为例)
在反应瓶中加入6a 5.85 g(10 mmol),碳酸钾6.50 g,DMF 100 mL 和水 30 mL,搅拌使其溶解;分批加入乙酰氧肟酸2.04 g(30 mmol),加毕,反应12 h。过滤,滤液浓缩至干后经柱色谱(洗脱剂:A=30∶1)纯化得白色固体7a 4.20 g。
用类似方法合成白色固体7b~7h。
7a:产率 70.2%;1H NMR δ:0.83(d,J=6.8 Hz,6H),2.32(m,1H),2.64(t,J=7.2 Hz,2H),3.71(s,3H),3.78(s,3H),3.94(q,J=7.2 Hz,2H),4.22(d,J=7.2 Hz,1H),4.58(d,J=4.4 Hz,2H),6.81 ~6.88(m,3H),7.10 ~ 7.19(m,2H),7.33 ~ 7.55(m,3H),7.64(d,J=8.4 Hz,2H),8.39(d,J=2.8 Hz,1H),8.56(br,2H);ESI-MS m/z:599.4{[M+H]+};HR-ESI-MS m/z:Calcd for C31H34N8O5{[M+H]+}599.660 2,found 599.661 7。
7b:产率 48.8%;1H NMR δ:2.62(t,J=7.2 Hz,2H),3.69(s,3H),3.78(s,3H),3.94(q,J=7.2 Hz,2H),4.19(d,J=7.2 Hz,2H),4.58(d,J=4.4 Hz,2H),6.81 ~6.88(m,3H),7.10 ~ 7.19(m,2H),7.33 ~ 7.55(m,3H),7.64(d,J=8.4 Hz,2H),8.39(d,J=2.8 Hz,1H),8.56(br,2H);ESI-MS m/z:557.3{[M+H]+};HR-ESI-MS m/z:Calcd for C28H28N8O5{[M+H]+}557.580 5,found 557.582 7。
7c:产率 72.5%;1H NMR δ:0.85(d,J=6.8 Hz,3H),2.62(t,J=7.2 Hz,2H),3.70(s,3H),3.77(s,3H),3.92(q,J=7.2 Hz,2H),4.21(d,J=7.2 Hz,1H),4.54(d,J=4.4 Hz,2H),6.81 ~6.88(m,3H),7.14 ~7.19(m,2H),7.33 ~7.52(m,3H),7.60(d,J=8.4 Hz,2H),8.37(d,J=2.8 Hz,1H),8.53(br,2H);ESI-MS m/z:571.1{[M+H]+};HR-ESI-MS m/z:Calcd for C29H30N8O5{[M+H]+}571.241 7,found 571.240 2。
7d:产率 44.0%;1H NMR δ:0.91(d,J=6.8 Hz,6H),1.56(m,1H),1.99(m,2H),2.64(t,J=7.2 Hz,2H),3.71(s,3H),3.78(s,3H),3.94(q,J=7.2 Hz,2H),4.42(d,J=7.2 Hz,1H),4.58(d,J=4.4 Hz,2H),6.81 ~6.88(m,3H),7.10 ~ 7.19(m,2H),7.33 ~7.55(m,3H),7.61(d,J=8.4 Hz,2H),8.22(d,J=2.8 Hz,1H),8.46(br,2H);ESIMS m/z:613.4{[M+H]+};HR-ESI-MS m/z:Calcd for C32H36N8O5{[M+H]+}613.686 8,found 613.684 6。
7e:产率 65.2%;1H NMR δ:0.86(d,J=6.8 Hz,6H),2.32(m,1H),2.66(t,J=7.2 Hz,2H),3.75(s,3H),3.79(s,3H),3.91(q,J=7.2 Hz,2H),4.22(d,J=7.2 Hz,1H),4.55(d,J=4.4 Hz,2H),6.81 ~6.88(m,3H),7.10 ~ 7.19(m,2H),7.34 ~ 7.55(m,3H),7.64(d,J=8.4 Hz,2H),8.35(d,J=2.8 Hz,1H),8.50(br,2H);ESI-MS m/z:600.3{[M+H]+};HR-ESI-MS m/z:Calcd for C31H33N7O6{[M+H]+}600.645 0,found 600.643 3。
7f:产率 36.9%;1H NMR δ:2.58(t,J=7.2 Hz,2H),3.65(s,3H),3.78(s,3H),3.91(q,J=7.2 Hz,2H),4.14(d,J=7.2 Hz,2H),4.55(d,J=4.4 Hz,2H),6.82 ~6.84(m,3H),7.13 ~ 7.17(m,2H),7.30 ~ 7.58(m,3H),7.61(d,J=8.4 Hz,2H),8.35(d,J=2.8 Hz,1H),8.50(br,2H);ESI-MS m/z:558.2{[M+H]+};HR-ESI-MS m/z:Calcd for C28H27N7O6{[M+H]+}558.565 2,found 558.564 5。
7g:产率 46.7%;1H NMR δ:0.86(d,J=6.8 Hz,3H),2.61(t,J=7.2 Hz,2H),3.73(s,3H),3.79(s,3H),3.92(q,J=7.2 Hz,2H),4.21(d,J=7.2Hz,1H),4.54(d,J=4.4 Hz,2H),6.81 ~6.88(m,3H),7.14 ~7.19(m,2H),7.33 ~7.52(m,3H),7.62(d,J=8.4 Hz,2H),8.31(d,J=2.8 Hz,1H),8.53(br,2H);ESI-MS m/z:572.3{[M+H]+};HR-ESI-MS m/z:Calcd for C29H29N7O6{[M+H]+}572.591 8,found 572.590 9。
7h:产率26.8%;1H NMR δ:0.94(d,J=6.8 Hz,6H),1.50(m,1H),1.92(m,2H),2.63(t,J=7.2 Hz,2H),3.76(s,3H),3.82(s,3H),3.93(q,J=7.2 Hz,2H),4.41(d,J=7.2 Hz,1H),4.52(d,J=4.4 Hz,2H),6.84 ~6.89(m,3H),7.12 ~7.17(m,2H),7.32 ~7.56(m,3H),7.66(d,J=8.4 Hz,2H),8.24(d,J=2.8 Hz,1H),8.48(br,2H);ESI-MS m/z:614.2{[M+H]+};HR-ESI-MS m/z:Calcd for C32H35N7O6{[M+H]+}614.671 5,found 614.670 8。
1.3 抗凝活性评价-活化部分凝血活酶时间的测定
将质量18 g~20 g的昆明小鼠,随机分组,每组10只,禁食过夜。将达比加群酯及7a~7h悬浮或溶解于1%的羧甲基纤维素钠水溶液中,配成1 mg·mL-1的浓度,按 10 mg·Kg-1的剂量(折合成达比加群计算)灌胃给药,30 min后通过心脏穿刺取血,加入4%枸杞酸钠溶液至0.4%终浓度抗凝,12 000 r·min-1离心 5 min。取血浆0.1 mL,加入 aPTT 试剂 0.1 mL,于 37 ℃ 预温 3 min,加入37℃氯化钙溶液0.1 mL,用血小板聚集凝血因子分析仪测定凝固时间,即为aPTT值。
2 结果与讨论
2.1 7 的合成
由于受到苯环对位杂原子的影响,7a~7d的产率明显高于7e~7h。这可能由于其对位的LINKER杂原子氧的电负性大于氮的电负性,造成了环合时氰基邻位的氟离去能力减弱,致使收率降低。其确切反应机理有待进一步研究。
2.2 抗凝血活性与结构的关系
7a~7h的抗凝血活性结果见表1。由表1可见,7d与达比加群酯的抗凝血活性基本相当;7h的活性比生理盐水的更弱,可能是因为结构中所连的氧桥连与亮氨酸修饰后影响了其体内的吸收。7c和7g的aPTT值虽然较高,但在临床上容易出现出血的风险;7a和7b既能延长凝血时间,又不至于出现出血的风险,因此活性较好;而7e和7f略比达比加群酯稍差,其原因可能在于与其相连的LINKER由氮替换为氧,使整个分子过于柔性,改变了其原有的空间大体骨架走向。在苯芳环与咪唑芳环所连接的LINKER需要采用略显刚性的氮桥连,可增强7对抗凝血活性,若连接过于柔性的氧桥连,则致使抗凝血活性明显降低,其构效关系有待进一步研究。
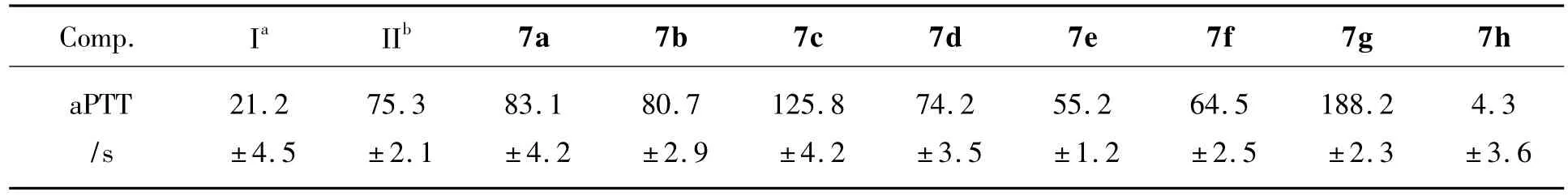
表1 7a~7h的抗凝活性Table 1 The inhibition activities of 7a~7h
3 结论
合成了8个新型的苯并咪唑类衍生物(7a~7h)。抗凝血活性实验结果表明:7a和7b的抗凝血活性最好,其aPTT值分别为(83.1±4.2)s和(80.7 ±2.9)s,优于对照药达比加群酯(75.3 ±2.1)s。
7a~7h的合成,对凝血抑制药物的开发具有重要意义。
[1]Blech S,Ebner T,Ludwing-Schwellinger E,et al.The metabolism and disposition of the oral direct thrombin inhibitors,dabigatran,in hunman[J].Drug Metab Dispos,2008,36(2):386 -399.
[2]Huber K,Connolly S J,Kher A,et al.Practical use of dabigatran etexilate for stroke prevention in atrial fibrillation[J].International Journal of Clinical Practice,2013,67(6):516 -526.
[3]Davidson T,Husberg M,Janzon M,et al.Cost-effectiveness of dabigatran compared with warfarin for patients with atrial fibrillation in Sweden[J].Eur Heart J,2013,34(3):177 -183.
[4]Gliem M,Hermsen D,Rooijen N,et al.Secondary intracerebral hemorrhage due to early initiation of oral anticoagulation after ischemic stroke:An experimental study in mice[J].Stroke,2012,43(12):3352 -3357.
[5]Norbert H H,Herbert N,Henning P,et al.Structurebased design of novel potent nonpeptide thrombin Inhibitors[J].J Med Chem,2002,45(9):1757 -1766.
[6]Obst U,Banner D W,Weber L,et al.Molecular recognition an the thrombin active site:Structure-based design and synthesis of potent and selective thrombin inhibitors and the X-ray crystal structures of two thrmbin-inhibitor complexes[J].Chem Biol,1997,4(4):287-295.
[7]Liu H Q,Zhang W G,Cai Z Q,et al.Dabigatran etexilate tetrahydrate[J].Acta Cryst E,2012,68:O3385.
[8]郭雅俊,朱雪焱,黄雨,等.达比加群酯的合成[J].合成化学,2014,22(2):262 -264.
[9]蔡志强,马维英,候旭,等.达比加群酯的合成工艺改进[J].精细化工,2015,32(3):308 -311.
[10] 张伟光,蔡志强,刘洪强,等.3-(吡啶-2-基氨基)丙酸乙酯的合成工艺研究[J].合成化学,2012,20(6):782-786.
