3,3-二氟环己基甲胺的简便合成*
2015-04-23赵玉灵仇记宽
赵玉灵,仇记宽
(河南师范大学化学化工学院精细化学品绿色制造河南省协同创新中心,河南新乡 453007)
环己基甲胺是重要的有机合成中间体,可用于医药、农药等的合成,如制备镇痛剂[1]、二肽半胱天冬酶抑制剂[2]、JNK 抑制剂[3]等。双氟代环己基甲胺作为环己基甲胺的衍生物,在医药及农药方面有着潜在的应用价值,其合成方法也是科学工作者研究的重点。该类化合物现有的合成方法存在诸多不足,如:步骤较多,后处理比较繁琐[4-5];需要用剧毒物质 KCN,安全系数低[4];滴加LiN(Pr-I)2需要低温,滴加完成后需加热回流,还原后有大量的副产物不宜分离[5];需要用氨基锂试剂,反应条件相对苛刻,且合成方法对3,3-二氟环己基甲胺(4)类衍生物具有一定的局限性,给产业化带来不便[6]等等。
本文报道一种合成4的新路线,即以环己烯酮为原料,与硝基乙酸乙酯经Michael加成制得2-硝基-2-(环己基3-酮)乙酸乙酯(1);1经水解脱羧制得3-硝基环己基酮(2);2与二乙胺基三氟化硫(DAST)经亲核氟化反应制得1,1-二氟-3-硝甲基-环己烷(3);3经氢化还原反应合成了4(Scheme 1),总收率55%,其结构经1H NMR和MS确证。并对还原反应条件进行了优化。

Scheme 1
1 实验部分
1.1 仪器与试剂
Bruker DPX-400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Bruker Esquire 3000型离子阱液相色谱质谱仪。
所用试剂均为分析纯。
1.2 合成
(1)1的合成
在三口瓶中依次加入乙腈250 mL和环己烯酮20.6 g(214.30 mmol),搅拌使其溶解;加入硝基乙酸乙酯30 g(225.39 mmol)和N,N-二异丙基乙胺(DIEA)29 g,氮气保护下于室温反应12 h。蒸除溶剂,残余物用乙酸乙酯200 mL溶解,用3 mol·L-1盐酸 (2 ×100 mL)洗涤,无水硫酸钠干燥,经硅胶柱层析[洗脱剂:A=V(石油醚)∶V(乙酸乙酯)=20∶1]纯化得淡黄色油状液体1 41.7 g,收率 85%;1H NMR δ:1.34(t,J=3.6 Hz,3H,CH3),1.59 ~ 1.75(m,2H,CH2),2.01 ~2.18(m,2H,CH2),2.24~2.50(m,4H,CH2),2.79 ~2.81(m,CH),4.27 ~4.36(m,2H,OCH2),5.01 ~5.08(m,1H,CHNO2);MS m/z:230{[M+H]+}。
(2)2的合成
在三口瓶中依次加入乙醇100 mL和1 20 g(87.25 mmol),搅拌使其溶解;加入 LiOH 10.9 g(259.77 mmol)的水(100 mL)溶液,于80 ℃反应4 h。蒸馏除溶剂,残余物用乙酸乙酯200 mL溶解后用3 mol·L-1盐酸洗涤,无水硫酸钠干燥,减压蒸除溶剂得淡黄色油状液体2 23.8 g,收率87%;1H NMR δ:1.47 ~ 1.55(m,1H,CH),1.68 ~1.99(m,2H,CH2),2.02~2.30(m,2H,CH2),2.32 ~2.64(m,4H,CH2),4.31 ~4.34(m,2H,CH2NO2);MS m/z:158{[M+H]+}。
(3)3的合成
在三口瓶中依次加入二氯甲烷200 mL和2 15 g(95.44 mmol),搅拌使其溶解;氮气保护下于0 ℃滴加 DAST 31.5 g(195.42 mmol),滴毕,于室温反应6 h。加入100 mL冰水萃灭反应,用二氯甲烷(2×100 mL)萃取,合并有机相,用无水硫酸钠干燥,减压蒸除溶剂,残留物经硅胶柱层析(洗脱剂:A=30∶1)纯化得淡黄色油状液体3 15.48 g,收率 91%;1H NMR δ:1.06 ~ 1.14(m,1H,CH),1.43 ~1.88(m,4H,CH2),2.03 ~2.52(m,4H,CH2),4.30(d,J=7.2 Hz,2H,NO2CH2);MS m/z:180{[M+H]+}。
(4)4的合成
在三口瓶中依次加入无水乙醇100 mL和3 15 g(83.73 mmol),搅拌使其溶解;加入Raney Ni 12 g,通入氢气,于室温反应12 h。过滤,滤液减压蒸除溶剂得黄色油状液体4 10.2 g,收率82%;1H NMR δ:1.02 ~ 1.15(m,1H,CH),1.41 ~2.15(m,8H,CH2),2.84 ~2.87(d,J=9.2 Hz,2H,CH2);13C NMR δ:122.15,46.68,40.54,40.21,38.16,31.67,28.98,13.44;MS m/z:150{[M+H]+}。
2 结果与讨论
2.1 合成4的反应条件优化
为提高4的总收率,重点对合成4的反应条件进行了优化。3 83.73 mmol,其它反应条件同1.2(4),考察了反应溶剂、催化剂种类及其用量对4收率的影响,结果见表1。
由表1 中 No.3和 No.5 ~No.8 可见,在其余反应条件相同的情况下,EtOH为溶剂时收率最高(78%)。最佳溶剂为EtOH。
由表1中No.1~No.4可见,以EtOH为溶剂时,收率随着Raney Ni用量的增加而增加,用量超过80%时,收率会降低。MS跟踪分析发现有其他副产物产生,所以催化剂最佳用量为80%。
此外,还对比了Raney Ni(80%)+NH3·H2O,Pd/C和Pt/C为催化剂对4收率的影响,结果见表1中 No.9~No.11。从中可见,以 Raney Ni(80%)+NH3·H2O为共催化剂时的收率最好(82%)。通过MS检测分析,Raney Ni还原3时会产生一定量的偶联副产物,该偶联产物极性和4的极性相近,难以分离纯化,加入催化量的氨水可以抑制偶联产物的生成。
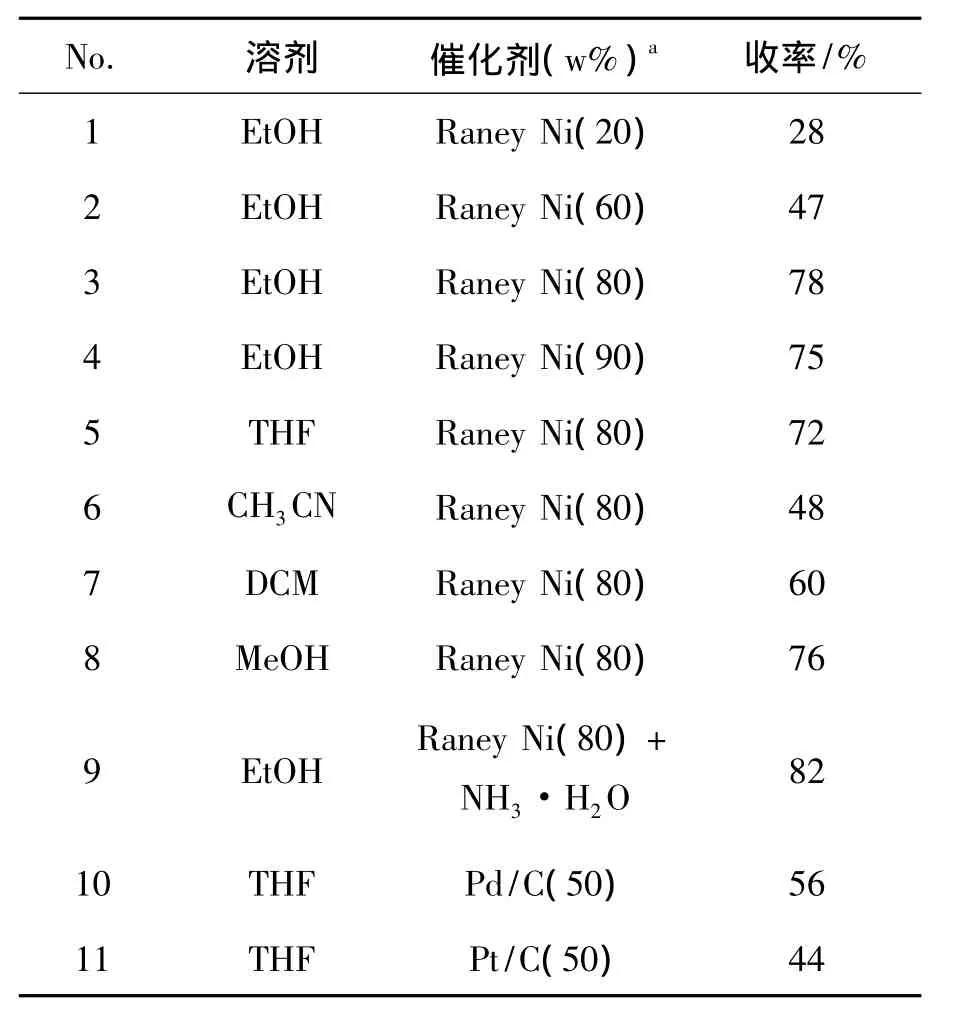
表1 溶剂及催化剂对反应收率的影响Table 1 Effect of solvent&reacion conditions on yield
综上所述,合成4的最佳反应条件为:1,1-二氟-3-硝甲基环己烷 83.73 mmol,Raney Ni(80%)和NH3·H2O为共催化剂,EtOH为溶剂,于室温反应12 h,收率82%。
3 结论
以环己酮为原料,通过Michael加成、水解脱羧、氟化和Raney Ni还原反应合成3,3-二氟环己基甲胺的方法为首次报道。该方法具有试剂廉价易得,反应条件温和,分离纯化容易,收率高等优点。为实验室制备环己基甲胺提供了一种新途径,也为合成环烷基甲胺类化合物提供了一种新方法,具有一定的工业化应用价值。
[1]William M L,Gerard G M P,Alan N,et al.Pyridine-3-carboxamides as novel CB2 agonists for analgesia[J].Bioorg Med Chem Lett,2009,19(1):259 -263.
[2]Hirokazu U,Makoto K,Hirohisa S,et al.Synthesis and structure-activity relationships of oxamyl dipeptide caspase inhibitors developed for the treatment of liver disease[J].Bioorg Med Chem Lett,2009,19(1):199 -202.
[3]Yasutomi A,Shuji K,Taiichi O,et al.Discovery,synthesis and biological evaluation of isoquinolones as novel and highly selective JNK inhibitor[J].Bioorg Med Chem,2008,16(8):4715 -4732.
[4]Chen L,Lin X,Qiu Z,et al.Compounds for the treatment and prevention of influenza[P].US WO 20 110 195 979 A1,2011.
[5]Chen X,Pierce B,Naing W,et al.Discovery of 2-chloro-N-[(4,4-difluoro-1-hydroxycyclohexyl)methyl]-5-(5-fluoropyrimidin-2-yl)benzamide as a potentand CNS penetrable P2X7 receptor antagonist[J].Bioorg Med Chem Lett,2010,20(10):3107 -3111.
[6]Mandal M,Blizzard T A,Chen H,et al.Preparation and use of bicyclic himbacine derivatives as par-receptor antagonists[P].US WO 2 013 134 012 A1,2013.
