CH3OH在Ga-rich GaAs(001)-(4×2)表面上的吸附与解离: 团簇模型的密度泛函计算
2015-03-23刘华成张建生
刘华成,张建生,于 锋
(西安工业大学理学院, 西安 710021)
CH3OH在Ga-rich GaAs(001)-(4×2)表面上的吸附与解离: 团簇模型的密度泛函计算
刘华成,张建生,于 锋
(西安工业大学理学院, 西安 710021)
采用简单团簇模型结合密度泛函理论研究了CH3OH在Ga-rich GaAs(001)-(4×2)表面上的吸附与解离过程. 计算结果表明, CH3OH在Ga-rich GaAs(001)-(4×2)表面上首先会形成两种化学吸附状态, 然后CH3OH经解离生成CH3O自由基和H原子吸附在表面不同位置上. 通过比较各个吸附解离路径, 发现解离后的H原子相对更容易吸附在位于表面第二层紧邻的As原子上.
Ga-rich GaAs(001)-(4×2)表面; CH3OH; 吸附; 团簇模型; 密度泛函理论
1 引 言
GaAs是一种非常常见的半导体材料, 该材料具有重要的应用价值; 具体来说, GaAs是性能优异的光阴极材料, 因而被广泛应用在微光夜视和加速器电子源的研制等领域[1,2].一些原子和分子容易吸附在GaAs表面上进而影响到GaAs材料的性能, 因此探索原子和分子在GaAs表面上的吸附一直是表面科学领域里的重要课题[3,4].
采用理论计算方法研究GaAs表面吸附问题时, 团簇模型一直扮演着重要的角色[4-18]. 团簇模型能够展现表面吸附问题的本质, 而且计算量相对较小. 在原子吸附方面, Ray研究组[5-11]进行了持续而深入的研究, 他们构建了不同大小的GaAs表面团簇模型, 系统研究了不同原子在不同位置上的吸附构型, 并给出了吸附能. Guo-Ping和Ruda[12]也采用团簇模型研究了S原子在GaAs(100)表面的吸附, 他们发现S原子倾向于吸附在桥位上并会显著影响表面的结构. 当分子吸附到GaAs表面时容易发生解离反应, 从而形成解离吸附状态. 在探索分子的吸附解离过程方面, 团簇模型也起了很大的作用. 根据团簇模型的计算结果, AsH3[4], NH3[13], CH3SH[14]和H2S[15-17]等分子在GaAs(001)-(4×2)表面上都容易发生H原子迁移. 相对来讲, C3H7SH分子的吸附解离更为复杂[18]. Tang和Cao[18]利用团簇模型详细研究了C3H7SH分子在Ga-rich GaAs(001)-(4×2)表面上发生吸附解离的反应路径, 并给出了详细的反应机理.
Büchel与Lüth[19]在超高真空条件下采用紫外光电子能谱研究了H2O和CH3OH分子在GaAs(110)表面上的吸附情况, 他们发现在表面上CH3OH会解离生成CH3O自由基和H原子. 在本论文中, 我们选取CH3OH分子作为研究对象, 采用团簇模型着重研究CH3OH分子在Ga-rich GaAs(001)-(4×2)表面上的吸附与解离反应机理. 通过计算期望回答如下三个问题: 第一, CH3OH分子在Ga-rich GaAs(001)-(4×2)表面上吸附以后是否容易发生解离? 第二, 解离主要沿着哪条反应路径? 第三, 解离机理和其它分子有何异同?
2 计算方法
采用团簇模型研究表面吸附问题时, 最为关键的一步是选择大小合适的团簇来模拟表面结构. 选择的标准是所选团簇能够反映表面的主要结构特征并且引入的计算量相对适中. 根据文献的结果[4,13,15-17], 我们选取Ga4As5H9团簇来模拟Ga-rich GaAs(001)-(4×2)表面. Lu等人[13]详细比较过Ga4As5H9和Ga7As8H11这两个团簇在模拟Ga-rich GaAs(001)-(4×2)表面时的差异. 根据他们的计算结果[13], 对于小分子吸附来讲, Ga4As5H9团簇已经可以较好地模拟Ga-rich GaAs(001)-(4×2)表面的结构特征. Ga4As5H9团簇中的H原子用于饱和悬挂键.
本论文中的计算工作均使用ORCA程序[20]完成. 在计算过程中采用广义梯度近似(GGA)泛函PBE[21,22], 基函数为def2-TZVPP基组[23,24]. 为了加快计算速度, 我们还利用了Resolution of the Identity (RI)近似[25], 辅助基函数为def2-TZVPP/J[26,27]. 另外, 本论文还采用色散校正的PBE-D3(BJ)泛函[28,29]进行了计算, 其中BJ代表Becke-Johnson damping[30-32]. 色散校正对合理描述吸附现象起着重要的作用, 它能够使密度泛函方法更加准确地计算能量和结构. 密度泛函计算过程中使用的积分网格为Grid4. 首先在PBE/def2-TZVPP和PBE-D3(BJ)/def2-TZVPP理论水平下分别优化了CH3OH分子和Ga4As5H9团簇相互作用势能面上化学吸附状态(Chemisorption State, CS), 过渡态(Transition State, TS)和解离吸附状态(Dissociation State, DS)的分子结构. 随后, 在同样的理论计算水平下通过数值频率分析确定了这些结构并获得了相应的零点能. 对于过渡态, 虚频振动模式表明了过渡态的连接属性. 本论文中的分子结构图采用MacMolPlt[33]程序绘制.
3 结果与讨论
3.1 CH3OH分子在Ga4As5H9团簇上的吸附解离路径
根据已发表文献的结果[4,13,15-17], 本论文采用Ga4As5H9团簇来模拟Ga-rich GaAs(001)-(4×2)表面. 在PBE/def2-TZVPP和PBE-D3(BJ)/def2-TZVPP理论水平下, Ga4As5H9团簇的优化结构基本一致. CH3OH分子与Ga4As5H9团簇相互作用势能面上关键点的分子结构见图1, 关键点的相对能量列于表1. 相应的反应势能剖面图如图2所示. 采用两种不同的计算水平(PBE/def2-TZVPP和PBE-D3(BJ)/def2-TZVPP)优化所得的分子结构参数略有差异, 具体的差异将在后面部分进行详细的讨论. 根据反应势能剖面图2, 吸附于Ga4As5H9团簇上CH3OH分子的O-H键将会断裂, 断键形成的H原子将与周围的Ga原子或As原子成键形成新的稳定结构, 发生H原子迁移所需要的能量各不相同. 通过计算发现, CH3OH在Ga4As5H9团簇上的吸附解离路径可能有三种, 详细过程列举如下:
路径I: CH3OH分子在Ga4As5H9团簇上首先形成化学吸附状态CS1, 随后经过渡态TS1形成解离吸附产物DS1. 对于解离吸附产物DS1, CH3O自由基吸附于Ga(1)原子上, H原子吸附在位于第二层的As(1)原子上. 这一路径可以简单记为CS1→TS1→DS1.
路径II: CH3OH分子在Ga4As5H9团簇上形成化学吸附状态CS2, 经过渡态TS1形成解离吸附状态DS2. 随后DS2经过渡态TS3形成解离吸附产物DS3. 其中DS3最显著的特点是O原子与第一层上的两个Ga原子成键形成Ga(1)-O-Ga(2)结构, 解离后的H原子与Ga(2)原子成键. 这一路径可以总结为CS2→TS2→DS2→TS3→DS3.
路径III: 由化学吸附状态CS2开始经过过渡态TS4生成解离吸附状态DS4, 这一反应路径和路径I类似. 对于解离吸附产物DS4, CH3O自由基吸附在位于第一层的Ga(1)原子上, 而解离后的H原子则吸附在位于第二层的As(2)原子上. 这一路径可以简单概括为CS2→TS4→DS4.
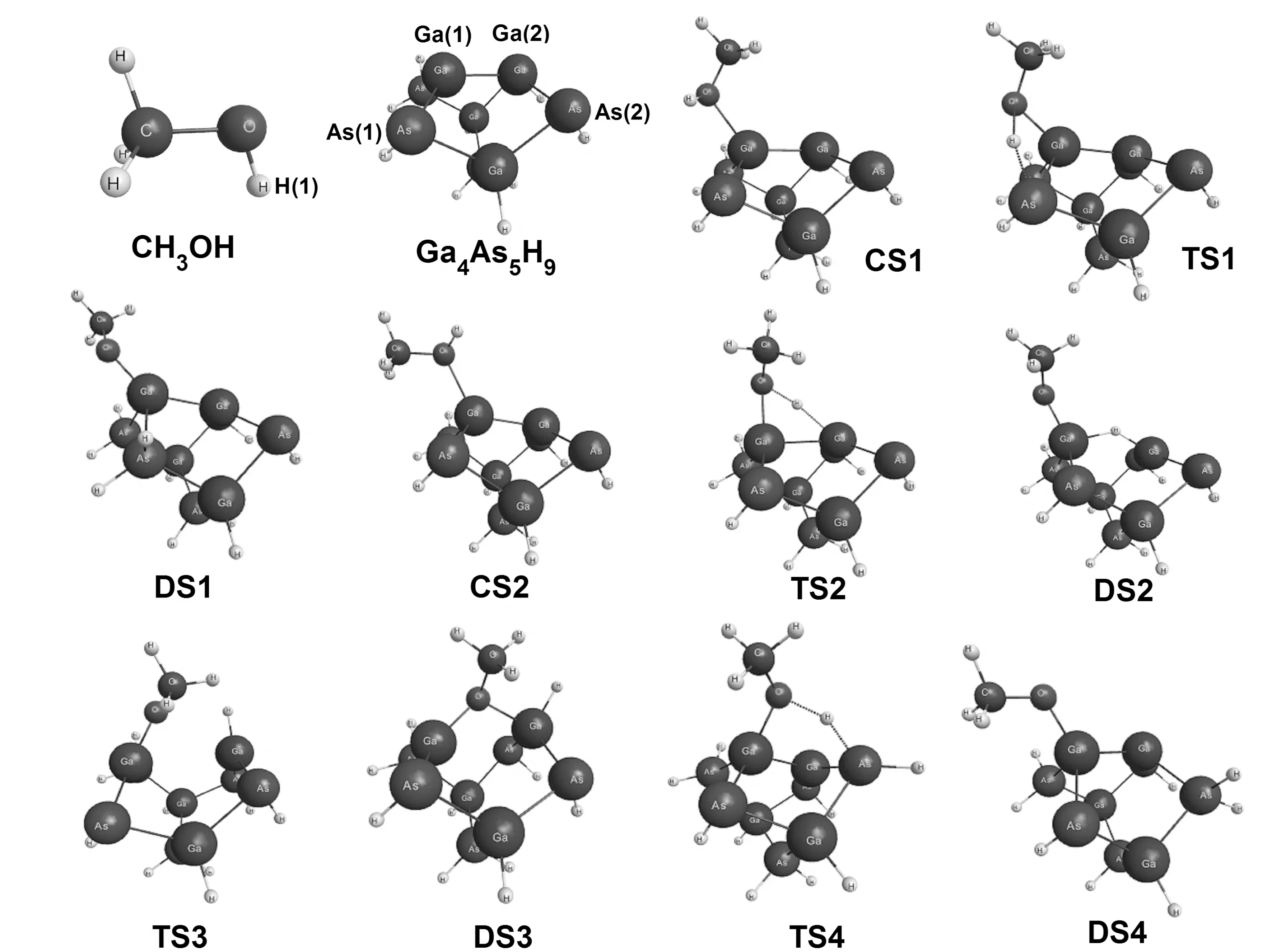
图1 CH3OH分子与Ga4As5H9团簇相互作用势能面上关键点的分子结构Fig. 1 Molecular geometries of the critical points on the potential energy surface for the interaction of CH3OH molecule with Ga4As5H9 cluster
表1 CH3OH分子与Ga4As5H9团簇相互作用势能面上关键点的相对能量 (单位:kJ·mol-1)
Table 1 Relative energies of the critical points on the potential energy surface for the interaction of CH3OH molecule with Ga4As5H9cluster (in kJ·mol-1)
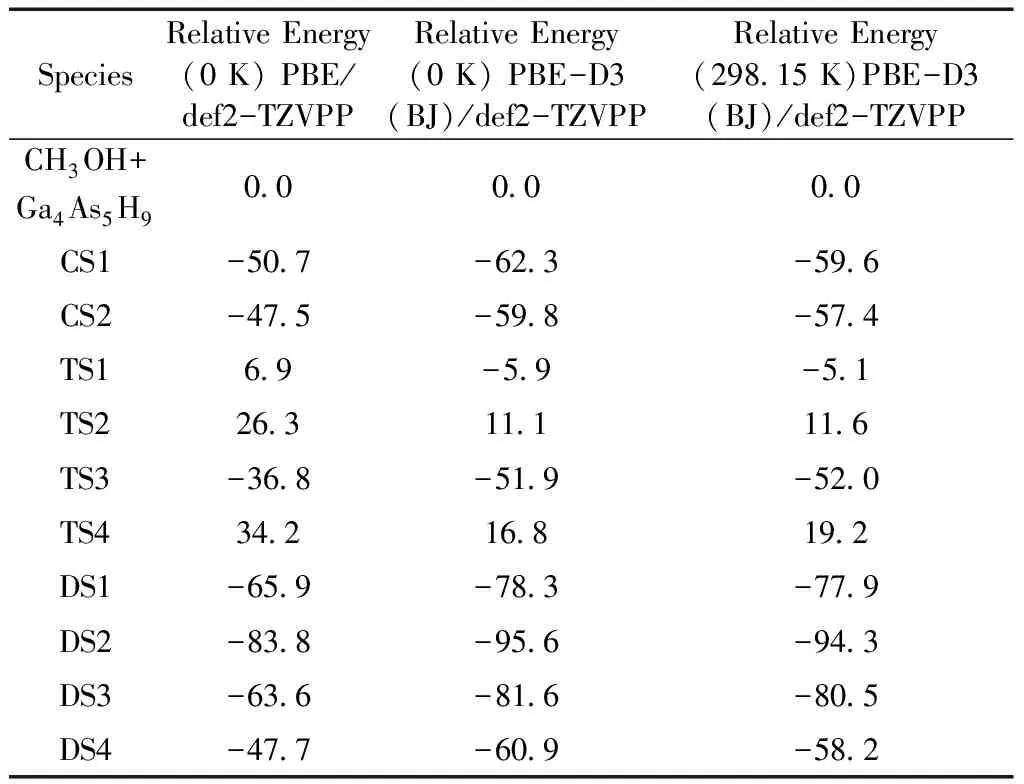
SpeciesRelativeEnergy(0K)PBE/def2-TZVPPRelativeEnergy(0K)PBE-D3(BJ)/def2-TZVPPRelativeEnergy(298 15K)PBE-D3(BJ)/def2-TZVPPCH3OH+Ga4As5H90 00 00 0CS1-50 7-62 3-59 6CS2-47 5-59 8-57 4TS16 9-5 9-5 1TS226 311 111 6TS3-36 8-51 9-52 0TS434 216 819 2DS1-65 9-78 3-77 9DS2-83 8-95 6-94 3DS3-63 6-81 6-80 5DS4-47 7-60 9-58 2
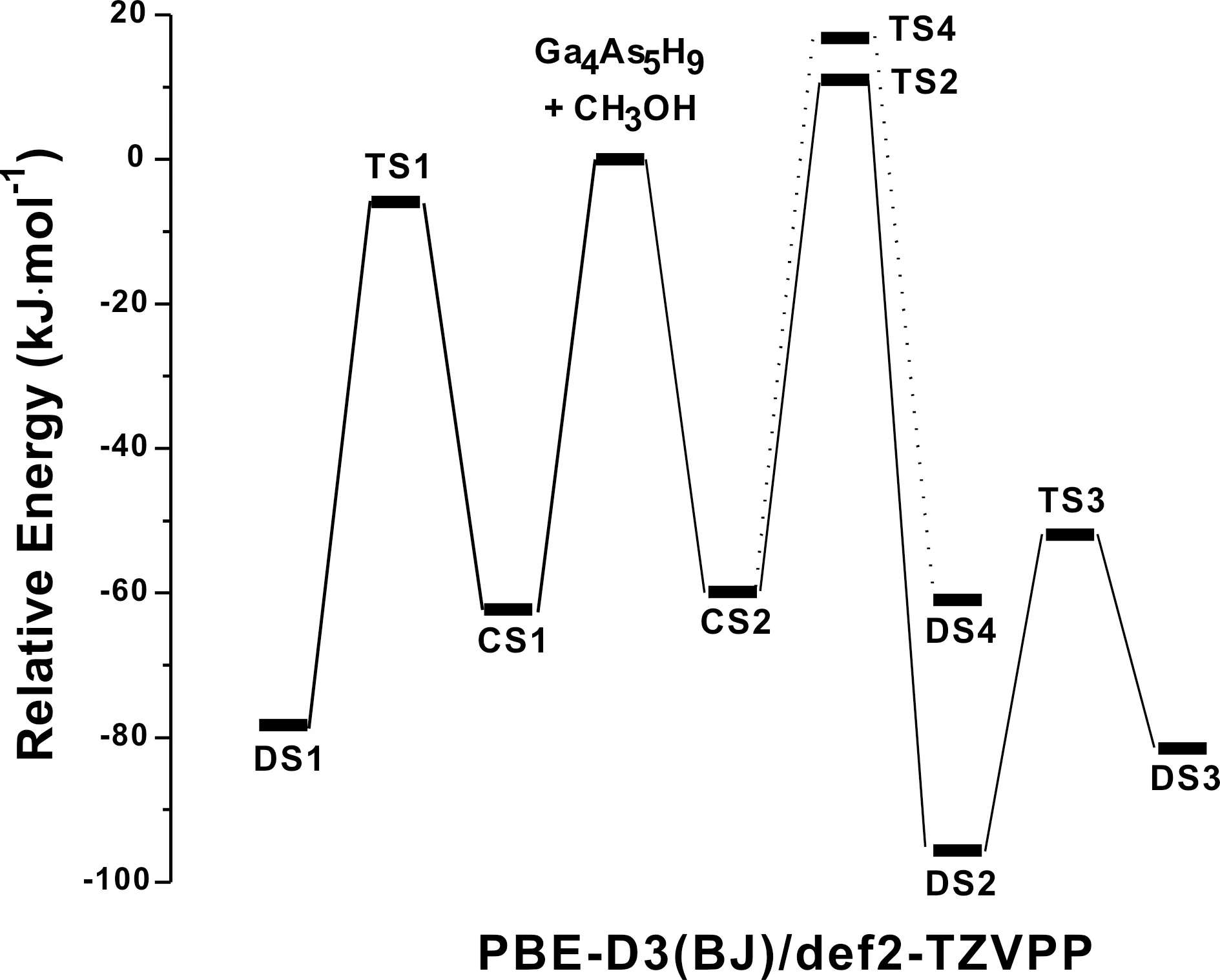
图2 CH3OH分子与Ga4As5H9团簇相互作用势能面 (0 K)Fig. 2 Potential energy profile for the interaction of CH3OH molecule with Ga4As5H9 cluster (0 K)
3.2 CH3OH分子在Ga4As5H9团簇上吸附解离过程中结构与能量的变化
在计算过程中分别采用了PBE泛函和色散校正的PBE-D3(BJ)泛函. 一般来说, 采用了色散校正的泛函更适合描述吸附解离过程[28,29]. 在下面的讨论中, 我们将对两种泛函的计算结果作一个对比. 根据反应路径上势垒的高低(如表1和图2所示), 路径I(CS1→TS1→DS1)是最容易发生的吸附解离过程. 路径III(CS2→TS4→DS4)的相对势垒最高, 沿该路径的吸附解离过程则难以发生.
CH3OH分子在Ga4As5H9团簇上首先会形成两种化学吸附状态(CS1和CS2). CS1和CS2的结构类似, 都是CH3OH分子中O原子与位于第一层的Ga(1)原子形成Ga(1)-O配位键. 不同之处在于吸附后CH3OH分子O-H键的指向不一样. 在PBE/def2-TZVPP理论水平下, CS1相对于反应物的能量为-50.7 kJ·mol-1(0 K), 其Ga(1)-O配位键的键长为2.197 Å; 而CS2相对于反应物的能量为-47.5 kJ·mol-1(0 K), 相应的Ga(1)-O配位键键长为2.215 Å. 在PBE-D3(BJ)/def2-TZVPP理论水平下, 结果略有不同. 具体来说, 在该理论水平下CS1相对于反应物的能量为-62.3 kJ·mol-1(0 K), 其Ga(1)-O配位键的键长为2.182 Å; 而CS2相对于反应物的能量为-59.8 kJ·mol-1(0 K), 相应的Ga(1)-O配位键键长为2.198 Å. 从上面的结果可以看出, PBE-D3(BJ)泛函计算得到的能量较低并且Ga(1)-O配位键的键长略短, 这是因为采用色散校正的PBE-D3(BJ)泛函考虑了相互作用能量中的色散能量部分[28,29], 更好地描述了化学吸附作用.
在下面的讨论过程中本论文将采用PBE-D3(BJ)泛函计算的结果. 对于吸附解离路径I, CS1需要越过渡态TS1生成解离吸附产物DS1. 在这个过程中H原子向紧邻的位于第二层的As(1)原子移动. O-H键的键长由CS1中的0.978 Å变为TS1中的1.480 Å. 最终O-H键断裂后H原子与位于第二层的As(1)原子成键形成产物DS1, 相应的As(1)-H键长为1.518 Å. 解离后的CH3O自由基吸附在第一层的Ga(1)原子上. Ga(1)-O键由CS1中的2.182 Å缩短为TS1中的1.938 Å, 最终形成的解离产物DS1的Ga(1)-O键为1.829 Å. 在PBE-D3(BJ)/def2-TZVPP水平下过渡态TS1相对于反应物的能量为-5.9 kJ·mol-1(0 K), 由CS1越过势垒TS1需要的能量为56.4 kJ·mol-1(0 K).
CS2对应吸附解离反应路径II和III. 对于路径II,CH3OH分子中的O-H键经由过渡态TS2断裂. 断键后H原子会插入位于表面第一层的Ga(1)-Ga(2)键, 形成吸附解离产物DS2. DS2具有Ga(1)-H-Ga(2)结构. DS2的Ga(1)-O键长为1.825 Å, Ga(1)-H-Ga(2)对应的键长和键角分别为1.816 Å (Ga(1)-H), 1.707 Å (Ga(2)-H)和136.81°. 过渡态TS2相对于反应物的能量为11.1 kJ·mol-1(0 K), 相对于CS2的能量则为70.9 kJ·mol-1(0 K). 随后, DS1的Ga(1)-H键会经过渡态TS3断裂形成DS3. 不同于DS2, DS3的结构特征是Ga(1)-O-Ga(2)结构. Ga(1)-O与Ga(2)-O的键长分别为1.891 Å和2.124 Å, Ga(1)-O-Ga(2)的键角为111.62°; Ga(2)-H的键长为1.592 Å. 过渡态TS3相对于反应物的能量为-51.9 kJ·mol-1(0 K), 相对于DS2的能量则为43.7 kJ·mol-1(0 K).
路径III和路径I相似, 区别在于路径III中H原子向第二层较远的As(2)原子迁移. 过渡态TS4相对于反应物的能量为16.8 kJ·mol-1(0 K), 相对于CS2的能量为76.6 kJ·mol-1(0 K). 因此, 相对来讲此解离过程难以发生.
3.3 与其它研究结果的比较
根据本论文的计算结果, CH3OH分子在Ga-rich GaAs(001)-(4×2)表面上很容易发生吸附和解离, 相应的解离产物为CH3O自由基和H原子. Buchel与Luth[19]在超高真空条件下采用紫外光电子能谱研究了CH3OH分子在GaAs(110)表面的吸附情况, 他们发现在GaAs(110)表面上CH3OH会解离生成CH3O自由基和H原子. 尽管本论文模拟的表面和他们实验研究的表面不一致, 但是CH3OH分子在吸附后解离生成的产物是一致的.
CH3OH分子在Ga-rich GaAs(001)-(4×2)表面上吸附解离的反应机理和AsH3[4], NH3[13],以及H2S[15-17]等分子的吸附解离机理相似. 尽管如此, 根据吸附解离路径上的势垒判断, CH3OH分子在Ga-Rich GaAs(001)-(4×2)表面上吸附后相对更容易发生解离(在PBE-D3(BJ)/def2-TZVPP理论水平下, TS1相对于反应物的能量为-5.9 kJ·mol-1). 此外, 本论文采用了和已发表文献[4,13,15-17]不同的计算方法, 首先是采用了色散校正的PBE-D3(BJ)泛函[28,29], 该泛函能够更好地描述吸附解离过程; 其次是运用了精度较高的Triple-ζ基函数def2-TZVPP[23,24].
4 结 论
在PBE/def2-TZVPP和PBE-D3(BJ)/def2-TZVPP理论水平下, 采用Ga4As5H9团簇模型研究了CH3OH分子在Ga-rich GaAs(001)-(4×2)表面上的吸附与解离机理. 根据本论文的计算结果, CH3OH分子在Ga-rich GaAs(001)-(4×2)表面上吸附后容易发生解离, 解离后的CH3O自由基吸附在位于表面第一层的Ga原子上, H原子相对更容易吸附在位于表面第二层紧邻Ga原子的As原子上.
[1] Zhang Y J, Chang B K, Yang Z,etal. Distribution of carriers in gradient-doping transmission-mode GaAs photocathodes grown by molecular beam epitaxy [J].Chin.Phys. B, 2009, 18(10): 4541.
[2] Zou J J, Zhang Y J, Yang Z,etal. Degradation model of GaAs vacuum electron sources [J].ActaPhys.Sin., 2011, 60(1): 017902 (in Chinese) [邹继军, 张益军, 杨智, 等. GaAs 真空电子源衰减模型研究 [J]. 物理学报, 2011, 60(1): 017902]
[3] Chung C H, Yi S I, Weinberg W H. Adsorption state of hydrogen sulfide on the GaAs (001)-(4× 2) surface [J].J.Vac.Sci.Technol. A, 1997, 15(3): 1163.
[4] Fu Q, Li L, Li C H,etal. Mechanism of arsine adsorption on the gallium-rich GaAs (001)-(4×2) surface [J].J.Phys.Chem. B, 2000, 104(23): 5595.
[5] Song K M, Ray A K.Abinitiostudy of cesium chemisorption on the GaAs (110) surface [J].Phys.Rev. B, 1994, 50(19): 14255.
[6] Song K M, Ray A K. Anabinitiostudy of potassium chemisorption on the GaAs (110) surface [J].J.Phys.:Condens.Matter, 1994, 6(45): 9571.
[7] Song K M, Khan D C, Ray A K. Correlation study of sodium-atom chemisorption on the GaAs (110) surface [J].Phys.Rev. B, 1994, 49(3): 1818.
[8] Song K M, Ray A K. A cluster study of Rb atom chemisorption on a GaAs (110) surface [J].J.Phys.:Condens.Matter, 1996, 8(36): 6617.
[9] Panda M, Ray A K. Density-functional cluster study of K adsorption on GaAs (110) surface [J].J.Vac.Sci.Technol. A, 1999, 17(5): 2647.
[10] Schailey R, Ray A K. An ab initio cluster study of chemisorption of atomic Cs on Ga-rich GaAs (100)(2×1),(2×2), and β (4×2) surfaces [J].J.Chem.Phys., 1999, 111: 8628.
[11] Mayo M L, Ray A K. A cluster study of aluminum adsorption on Ga-rich GaAs (100)(2×1) and β (4×2) surfaces [J].Eur.Phys.J. D, 2005, 33(3): 413.
[12] Guo-Ping J, Ruda H E.Abinitiostudies of S chemisorption on GaAs (100) [J].J.Appl.Phys., 1996, 79(7): 3758.
[13] Lu H L, Chen W, Ding S J,etal. DFT calculations of NH3adsorption and dissociation on gallium-rich GaAs (001)-4×2 surface [J].Chem.Phys.Lett., 2007, 445(4): 188.
[14] Lebedev M V. Methylthiol adsorption on GaAs (100)-(2×4) surface: Ab initio quantum-chemical analysis [J].Semiconductors, 2008, 42(9): 1048.
[15] Lu H L, Chen W, Ding S J,etal. Quantum chemical study of adsorption and dissociation of H2S on the gallium-rich GaAs (001)-4×2 surface [J].J.Phys.Chem. B, 2006, 110(19): 9529.
[16] Lebedev M V. Mechanism of H2S molecule adsorption on the GaAs (100) surface: Ab initio quantum-chemical analysis [J].Phys.SolidState, 2006, 48(1): 164.
[17] Shi M H.UsingdensityfunctionaltheorytostudythereactionmechanismofHS-R (R=H, CxH2x+1, x=1~8)ontheGa-richGaAs(100)surface[D]. China: National Cheng Kung University, 2007(in Chinese)[施旻宏. 利用密度泛函理论来研究HS-R (R=H, CxH2x+1, x=1~8)在Ga-Rich GaAs(100)表面上的反应机制[D]. 中国: 国立成功大学, 2007]
[18] Tang S, Cao Z. Density functional characterization of adsorption and decomposition of 1-propanethiol on the Ga-rich GaAs (001) surface [J].J.Phys.Chem. A, 2009, 113(19): 5685.
[19] Büchel M, Lüth H. Adsorption of water and methanol on GaAs (110) surfaces studied by ultraviolet photoemission [J].Surf.Sci., 1979, 87(2): 285.
[20] Neese F. The ORCA program system [J].WIREsComput.Mol.Sci., 2012, 2(1): 73.
[21] Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple [J].Phys.Rev.Lett., 1996, 77(18): 3865.
[22] Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)] [J].Phys.Rev.Lett., 1997, 78(7): 1396.
[23] Schäfer A, Horn H, Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr [J].J.Chem.Phys., 1992, 97(4): 2571.
[24] Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy [J].Phys.Chem.Chem.Phys., 2005, 7(18): 3297.
[25] Neese F. An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix [J].J.Comp.Chem., 2003, 24(14): 1740.
[26] Eichkorn K, Weigend F, Treutler O,etal. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials [J].Theor.Chem.Acc., 1997, 97(1-4): 119.
[27] Weigend F. Accurate Coulomb-fitting basis sets for H to Rn [J].Phys.Chem.Chem.Phys., 2006, 8(9): 1057.
[28] Grimme S, Antony J, Ehrlich S,etal. A consistent and accurateabinitioparameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu [J].J.Chem.Phys., 2010, 132(15): 154104.
[29] Grimme S, Ehrlich S, Goerigk L. Effect of the damping function in dispersion corrected density functional theory [J].J.Comp.Chem., 2011, 32(7): 1456.
[30] Becke A D, Johnson E R. Exchange-hole dipole moment and the dispersion interaction [J].J.Chem.Phys., 2005, 122(15): 154104.
[31] Johnson E R, Becke A D. A post-Hartree-Fock model of intermolecular interactions [J].J.Chem.Phys., 2005, 123(2): 024101.
[32] Johnson E R, Becke A D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections [J].J.Chem.Phys., 2006, 124(17): 174104.
[33] Bode B M, Gordon M S. MacMolPlt: a graphical user interface for GAMESS [J].J.Mol.GraphicsModel., 1998, 16(3): 133.
Adsorption and dissociation of CH3OH on the Ga-rich GaAs(001)-(4×2) surface: DFT computations with a cluster model
LIU Hua-Cheng,ZHANG Jian-Sheng,YU Feng
(School of Science, Xi'an Technological University, Xi'an 710021, China)
Based on a simple cluster model, the adsorption and dissociation processes of CH3OH molecule on the Ga-rich GaAs(001)-(4×2) surface have been studied with density functional theory. The present computations demonstrate that two chemisorbed states are formed initially, and then, the CH3OH molecule will decompose to CH3O radical and H atom on the surface. By comparing different adsorption and decomposition pathways, it is found that the dissociated H atom tends to be chemisorbed on the adjacent As atom at the second layer.
Ga-rich GaAs(001)-(4×2) surface; CH3OH; Adsorption; Cluster model; Density functional theory
2014-01-18
刘华成(1988—),男,硕士研究生,新疆石河子人,主要从事固体表面吸附计算研究.E-mail: huac.liu@gmail.com
于锋.E-mail: fengyu@xatu.edu.cn
103969/j.issn.1000-0364.2015.08.011
O641
A
1000-0364(2015)08-0591-06
