外电场对2-氟-5-溴吡啶分子结构与电子光谱影响的研究
2015-03-23吴学科梁冬梅
吴学科, 张 颂, 梁冬梅,2
(1.凯里学院物理与电子工程学院,凯里 556011; 2.山东大学化学与化工学院,济南 250100)
外电场对2-氟-5-溴吡啶分子结构与电子光谱影响的研究
吴学科1, 张 颂1, 梁冬梅1,2
(1.凯里学院物理与电子工程学院,凯里 556011; 2.山东大学化学与化工学院,济南 250100)
采用密度泛函B3LYP方法,在6-311++g(3df,3pd)基组水平上优化了不同外电场下2-氟-5-溴吡啶分子的基态稳定构型、电偶极矩和分子的总能量,在此基础上利用杂化CIS方法研究了外电场下2-氟-5-溴吡啶分子的前9个激发态的激发能、波长和振子强度受外电场的影响规律. 结果表明:5C-9Br和3C-10F间键长受到X轴向外场影响最大,随着外电场的继续增加,可能最先趋于断裂;在外电场F=-0.05 a.u.时总能量达到最大,而偶极矩达到最小;LUMO能级受外场影响较大,HOMO能级受外电场影响较小;激发波长、振子强度也受外电场影响,但随电场变化比较复杂.
2-氟-5-溴吡啶分子; 激发态; 外电场
1 引 言
吡啶及其众多衍生物是精细化工合成的重要中间体,在医药、农药、染料等领域有广泛应用,吡啶替代苯环还可以得到的具有更高生物活性或更低毒性新化合物,因此人们对吡啶及其众多衍生物进行了大量研究. 郭勇等采用密度泛函理论对2-氟-5-溴吡啶分子振动光谱进行了研究,并对分子的振动基频进行了理论归属[1],陈海群等介绍了2-氟吡啶在药物合成、生物技术、功能材料等方面的用途及其合成方法[2],方永勤等对吡啶衍生物的合成进行了研究[3-5]. 但从目前的文献来看,对2-氟-5-溴吡啶在外场作用下的激发态特性研究还未见报道,分子在外电场作用下会产生许多新的化学变化和现象,如新自由基的产生、化学键的断裂降解和新激发态的出现等,故分子外场效应的研究引起了许多学者的极大兴趣和重视[6,7],掌握2-氟-5-溴吡啶分子在一定外场作用下的激发规律,对于深入研究与2-氟-5-溴吡啶相关的药物具有重要的意义. 2-氟-5-溴吡啶分子在无外电场情况下的几何结构如图1所示,本工作采用密度泛函理论B3LYP方法在6-311++g(3df,3pd)基组水平上,对2-氟-5-溴吡啶分子沿X轴方向依次加入了-0.03 a.u.—0.03 a.u.强度的外电场进行几何结构优化,然后采用了杂化CIS方法,研究了外场下2-氟-5-溴吡啶分子的激发能,激发波长和振子强度. 其中1 a.u.=5.14225×1011V/m.
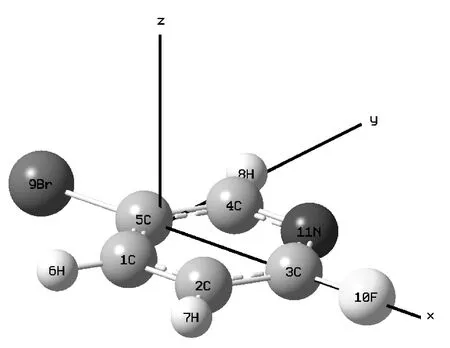
图1 无外电场作用下2-氟-5-溴吡啶分子结构Fig. 1 Optimized ground state geometry for 2-fluoro-5-bromopyridine molecule without electric field
2 理论和计算方法
外电场作用下分子体系的哈密顿量H为[8]:
H=H0+Hint
(1)
Hint代表辐射场与物质相互作用对原哈密顿量的附加项,也就是微扰项,在偶极近似下,分子体系与外电场F的相互作用项为:
Hint=-μ·F
(2)
μ为电偶极矩. 根据Grozema等提出的模型[9,10],对2-氟-5-溴吡啶分子沿X轴方向加强度依次为-0.03 a.u.—0.03 a.u.的外电场,对其分子结构进行优化,并在此基础上采用CIS方法对2-氟-5-溴吡啶分子的激发能、波长及振子强度等进行了计算.
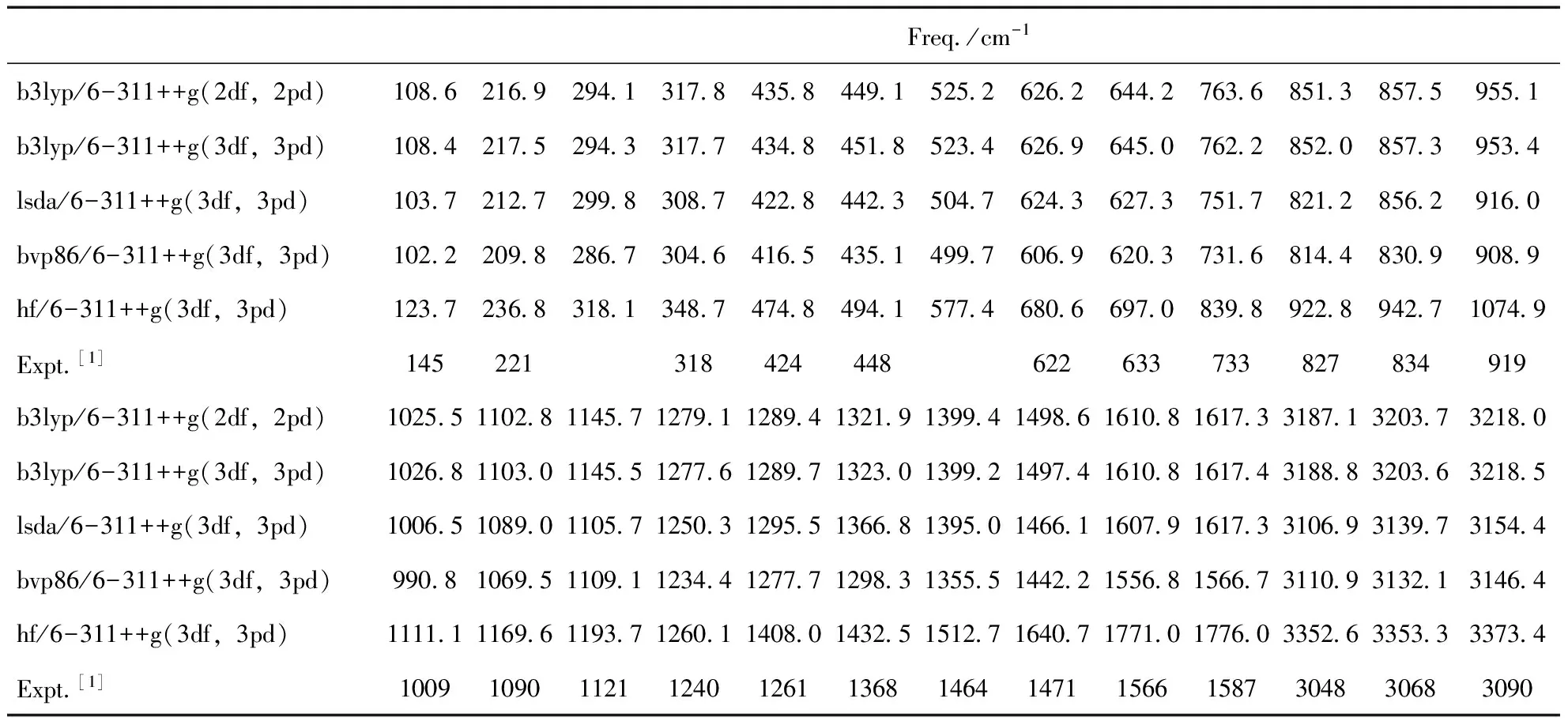
表1 2-氟-5-溴吡啶分子计算谐振频率与实验谐振频率的比较
3 计算结果与讨论
3.1 外电场下2-氟-5-溴吡啶分子基态稳定构型
无外场作用情况下,采用B3LYP、LSDA、BVP86、HF等方法对2-氟-5-溴吡啶分子进行优化计算,得出分子谐振频率如表1所示,可以看出,B3LYP/6-311++g(3df,3pd)方法优化得到的2-氟-5-溴吡啶分子谐振频率与实验值[1]符合较好,因此,在后面的计算选用该方法和基组来进行是可行的.
外电场(-0.03—0.03 a.u.)作用下,2-氟-5-溴吡啶分子优化后的几何参数、分子总能量、电偶极矩、轨道能量分别列在表2—4中.
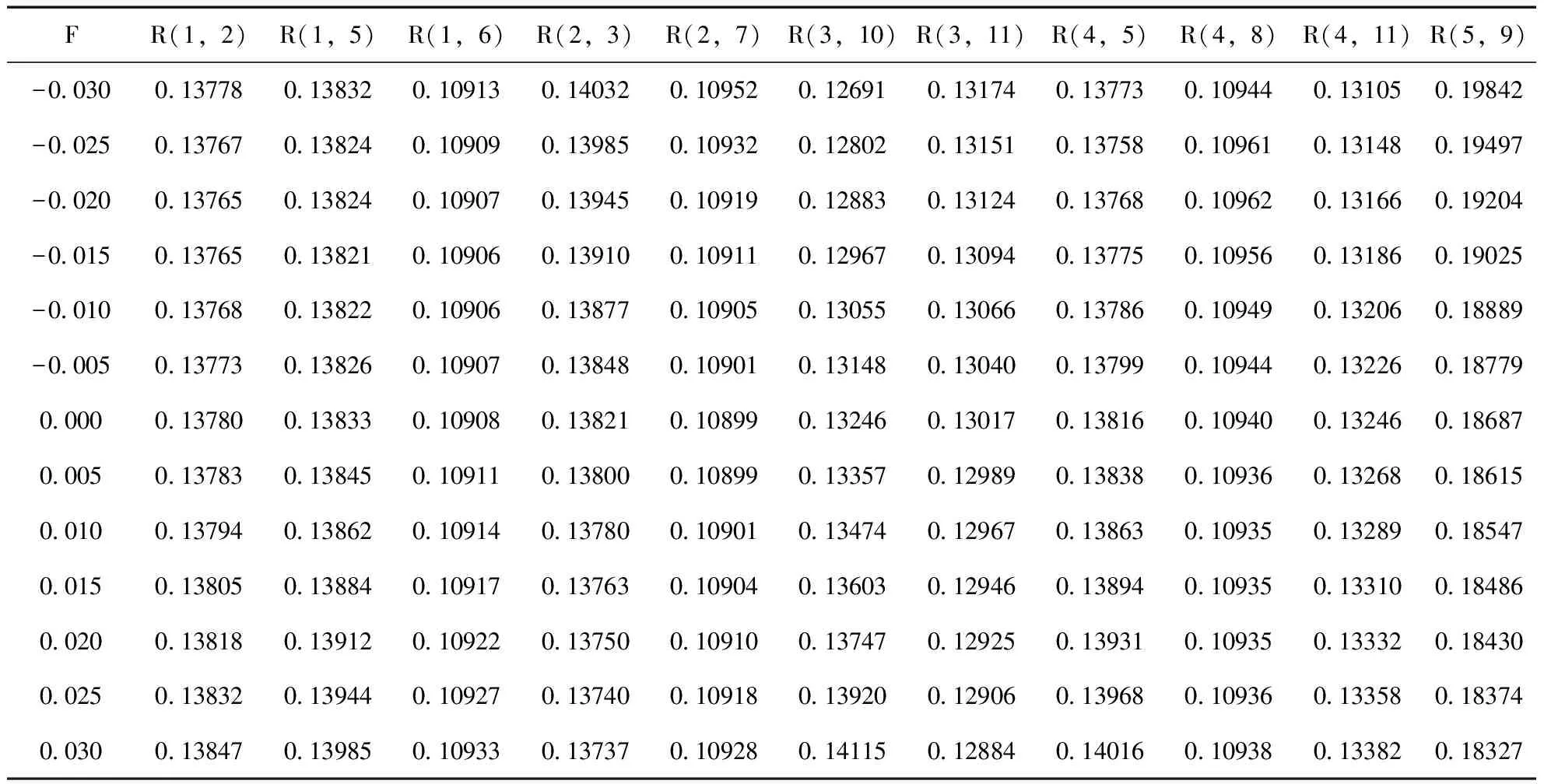
表2 外电场F(a.u.)下2-氟-5-溴吡啶分子的基态键长R(nm)
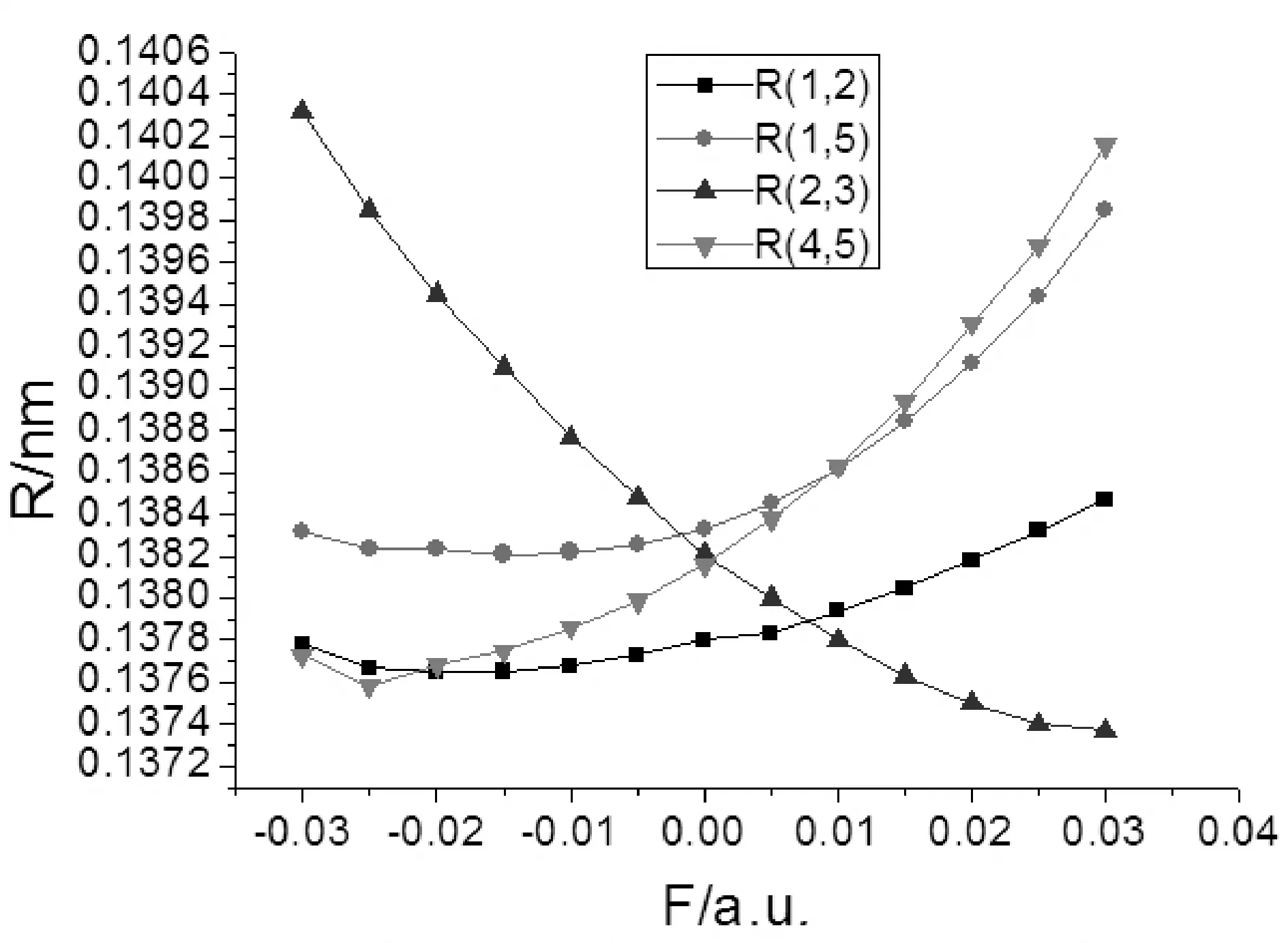
图2 2-氟-5-溴吡啶分子键长RC-C随外电场的变化Fig. 2 The variations of C-C bond distances in external electric fields for 2-fluoro-5-bromopyridine molecule
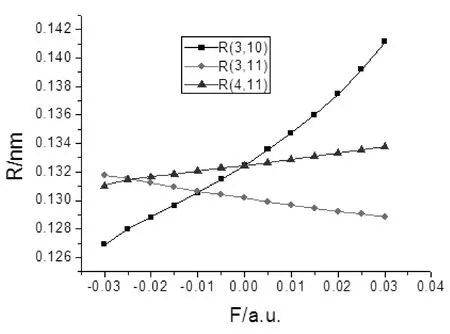
图3 2-氟-5-溴吡啶分子键长RC-F, RC-N随外电场的变化Fig. 3 The variations of C-F and C-N bond distances in external electric fields for 2-fluoro-5-bromopyridine molecule
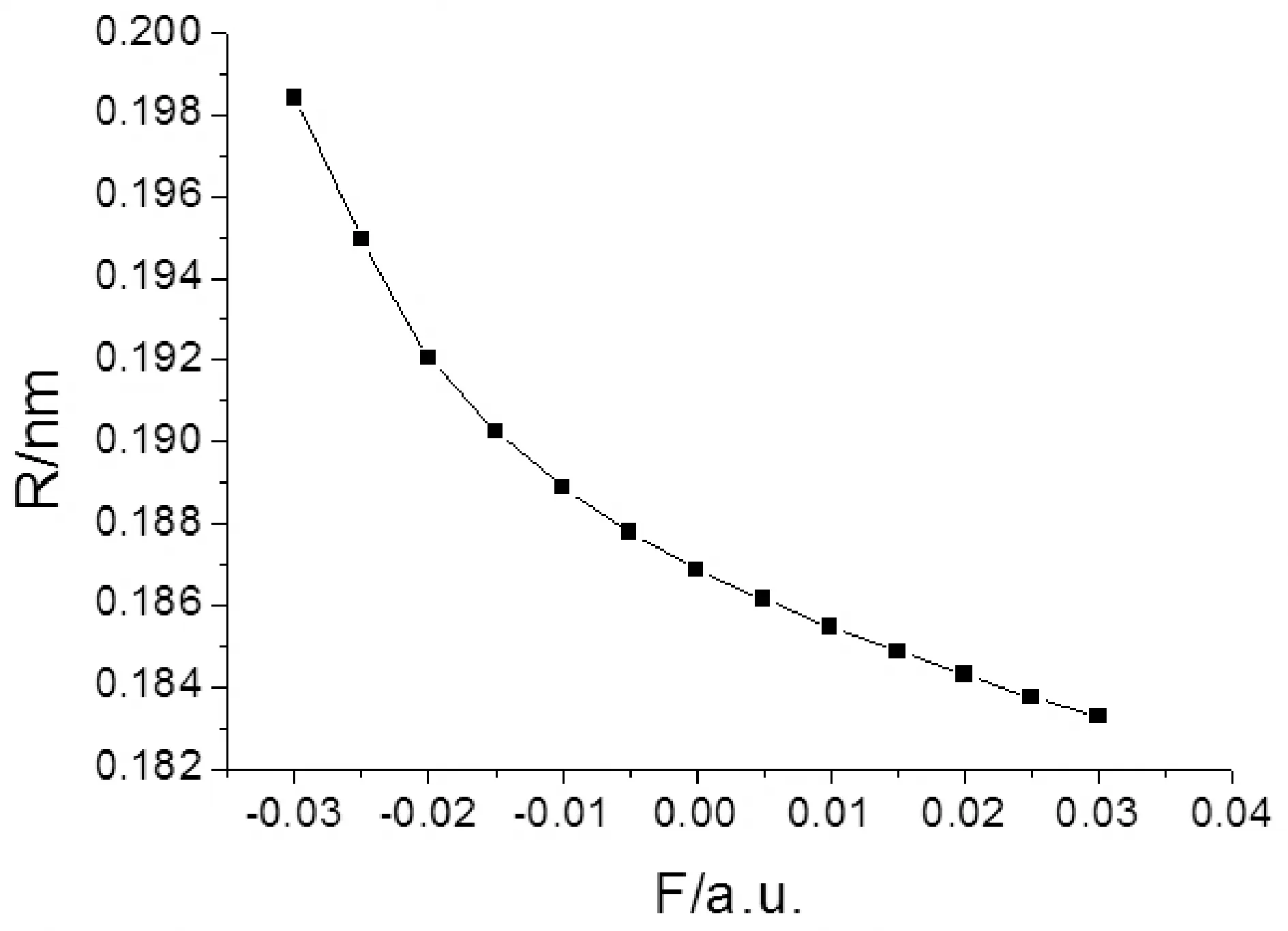
图4 2-氟-5-溴吡啶分子键长RC-Br随外电场的变化Fig. 4 The variation of C-Br bond distance in external electric fields for 2-fluoro-5-bromopyridine molecule
由表2和图2-4可以看出,2-氟-5-溴吡啶分子几何参数与外电场的大小有明显的依赖关系. 随着外电场强度的增大,R(3,10)和R(4,11)的键长增加,R(2,3)、R(3,11)和R(5,9)的键长减小,R(1,2)、R(1,5)和R(4,5)键长先减小再增加. 分子的这些几何参数变化可以用电荷转移引起分子内电场的变化来解释[11],在-0.03—0.03 a.u.范围外电场内,随着外电场增加,电子的局域转移导致3C-10F、4C-11N间的内电场减小,R(3,10)和R(4,11))的键长增加;而2C-3C、3C-11N和5C-9Br间的内电场增大,R(2,3)、R(3,11)和R(5,9)的键长减小;1C-2C、1C-5C和4C-5C的电子转移使得内电场先减小,但此时的内应力仍然大于外场力,随电场的继续增大,外场力大于内应力,故键长先减小再增加. 可以看出,C-H键长受外场影响最小,C-C键长受外场影响次之,5C-9Br和3C-10F间键长受到X轴向外场影响最大,可以预见,随着外电场的继续增加,最先趋于断裂的可能是5C-9Br键和3C-10F键.
3.2 外电场对2-氟-5-溴吡啶分子总能量、偶极矩和前线轨道能量的影响
2-氟-5-溴吡啶分子的总能量、偶极矩和前线轨道能量受外场作用的影响情况如表3和图5-7所示,可以看出,随外电场增大,分子总能量是先增大再减小,在外电场F=-0.005 a.u.时达到最大,而偶极矩先减小再增大,在外电场F=-0.005 a.u. 时达到最小. 最低空轨道能量EL受外场影响较大,当电场由-0.03 a.u.变化到-0.01 a.u.时,最低空轨道能量EL快速增加,由-0.01 a.u.变化到0.02 a.u.时,开始缓慢减小,而由0.02 a.u.变化到0.03 a.u.时,又快速减小;最高已占据轨道能量EH总体受外场影响较小,能隙EG受到外场作用时发生类似于最低空轨道能量EL的变化,当电场在-0.01— 0.02 a.u.范围变化时,能隙EG较大,变化缓慢,当外加0.02— 0.03 a.u.的正向电场、-0.01—-0.03 a.u.的负向电场时,能隙EG急剧下降.根据前线轨道理论[12],说明2-氟-5-溴吡啶分子给电子的能力受外电场影响较小,而接受电子能力和电子由占据轨道向低空轨道跃迁的能力受外电场影响较大,随着正负两个方向电场的增大,分子接受电子能力变得越强,电子很容易的由占据轨道向低空轨道跃迁.
表3 外电场下2-氟-5-溴吡啶分子的基态总能量E,偶极矩μ, 高占据轨道能量EH、最低空轨道能EL和能隙EG
Table 3 The total energy, dipole moment, HOMO energy, LUMO energy and energy gap of 2-fluoro-5-bromopyridine in external electric fields
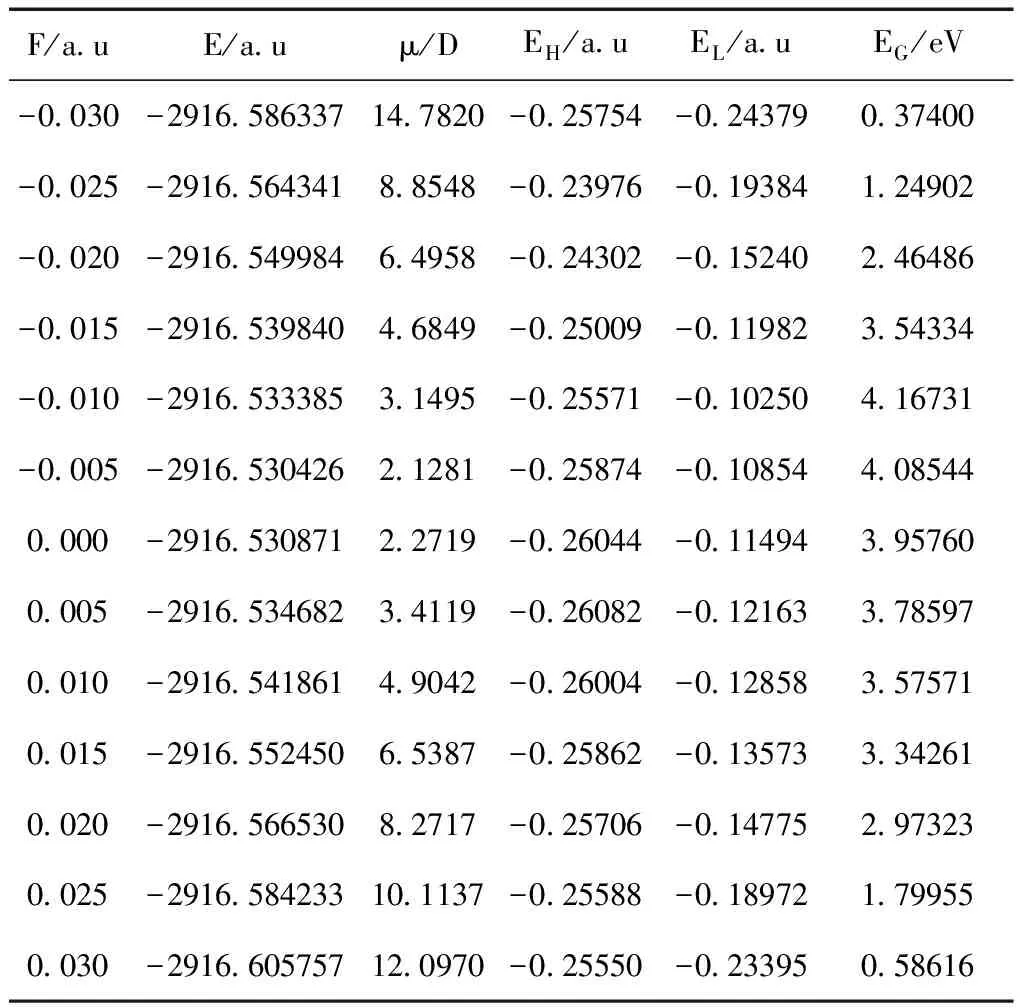
F/a uE/a uμ/DEH/a uEL/a uEG/eV-0 030-2916 58633714 7820-0 25754-0 243790 37400-0 025-2916 5643418 8548-0 23976-0 193841 24902-0 020-2916 5499846 4958-0 24302-0 152402 46486-0 015-2916 5398404 6849-0 25009-0 119823 54334-0 010-2916 5333853 1495-0 25571-0 102504 16731-0 005-2916 5304262 1281-0 25874-0 108544 085440 000-2916 5308712 2719-0 26044-0 114943 957600 005-2916 5346823 4119-0 26082-0 121633 785970 010-2916 5418614 9042-0 26004-0 128583 575710 015-2916 5524506 5387-0 25862-0 135733 342610 020-2916 5665308 2717-0 25706-0 147752 973230 025-2916 58423310 1137-0 25588-0 189721 799550 030-2916 60575712 0970-0 25550-0 233950 58616
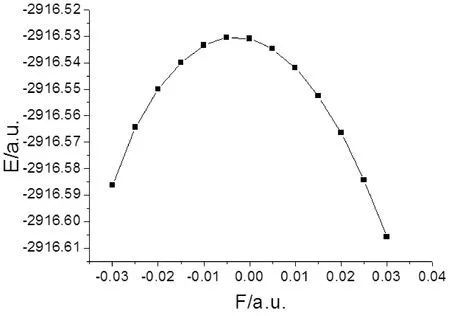
图5 2-氟-5-溴吡啶分子总能量随外电场的变化Fig. 5 The variation of total energy in external electric fields for 2-fluoro-5-bromopyridine molecule
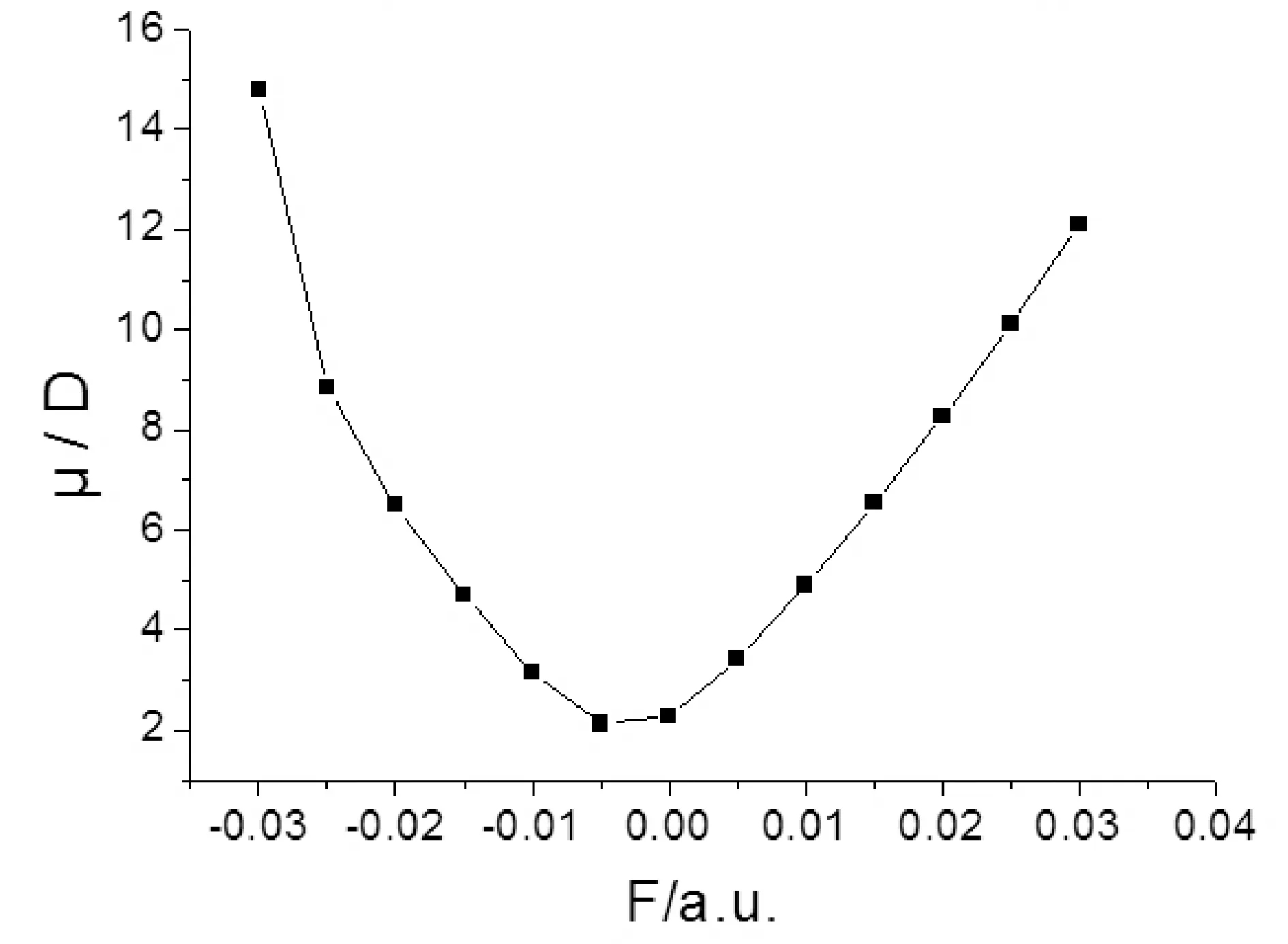
图6 2-氟-5-溴吡啶分子偶极矩随外电场强度的变化Fig. 6 The variation of dipole moment in external electric fields for 2-fluoro-5-bromopyridine molecule
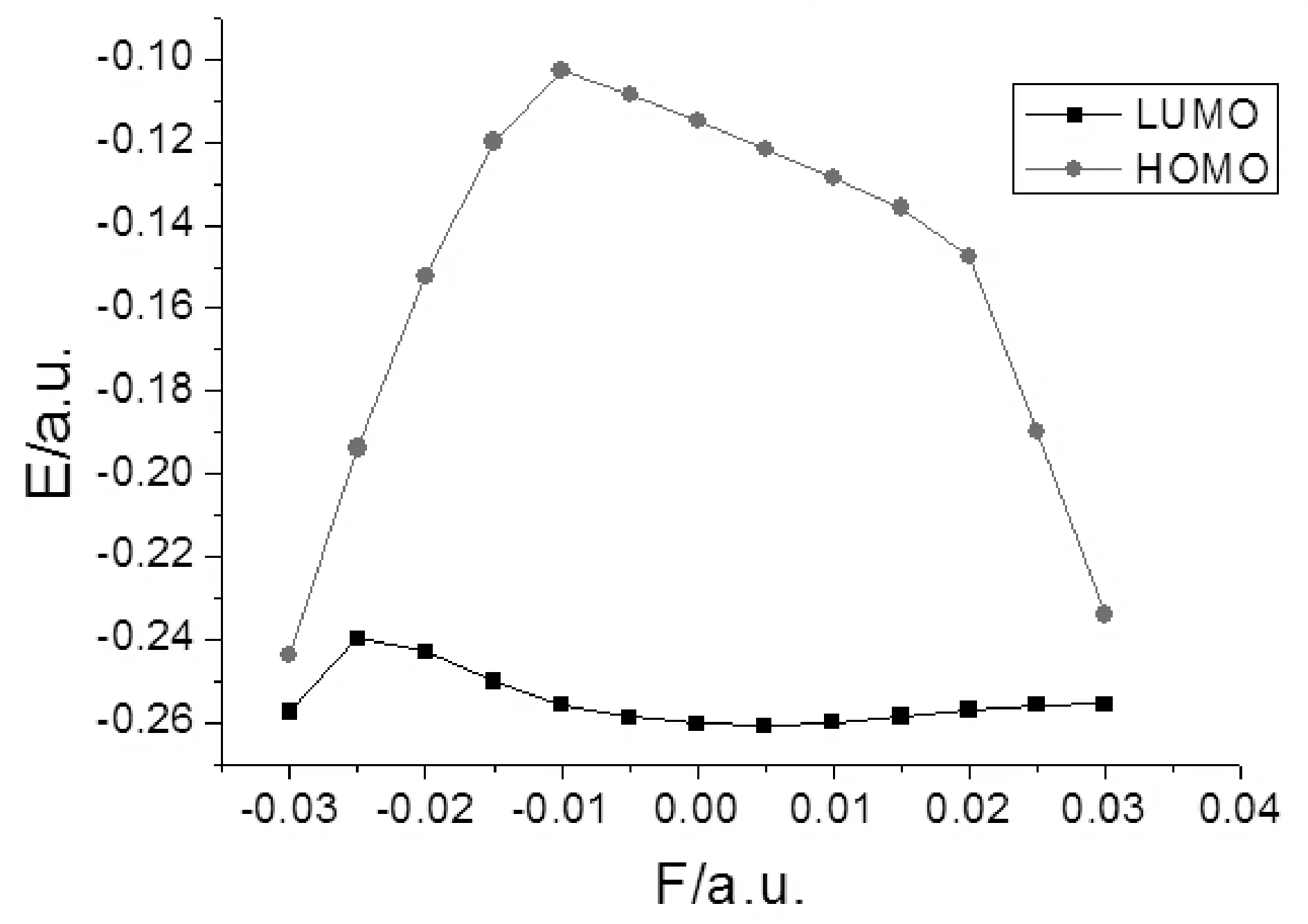
图7 2-氟-5-溴吡啶分子高占据轨道能量EH、最低空轨道能EL随外电场强度的变化Fig. 7 The variations of HOMO energy and LUMO energy in external electric fields for 2-fluoro-5-bromopyridine molecule
3.3 外电场对2-氟-5-溴吡啶分子激发态的影响
在2-氟-5-溴吡啶分子基态结构优化好的基础上,采用杂化CIS/6-311++g(3df,3pd)方法研究不同外电场下(0-0.03 a.u.)对2-氟-5-溴吡啶分子前9个激发态的激发波长、振子强度和紫外可见吸收光谱的影响, 如图8和表4所示,可以看出,在无电场时, 2-氟-5-溴吡啶分子在191.1 nm和207.6 nm处出现双紫外吸收峰,吸收强度分别为5037.06 L·mol-1·cm-1和5997.50 L·mol-1·cm-1,这主要是卤族原子取代的原因, 2-氟-5-溴吡啶分子的紫外吸收波段相对吡啶分子的紫外吸收波段发生了蓝移,同时吸收强度ε值减小. 当外电场强度的增大, 2-氟-5-溴吡啶分子的紫外光谱吸收峰发生红移,这由于是助色团-Br的孤电子和杂环上的π电子形成P-π共轭体系,π—π*跃迁需要的能量降低, 吸收峰出现了红移.
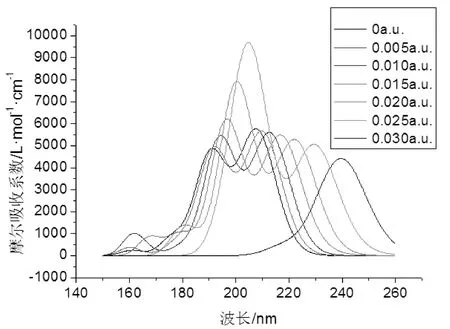
图8 2-氟-5-溴吡啶分子的紫外-可见吸收光谱Fig. 8 The UV-VIS absorption spectra of 2-fluoro-5-bromopyridine molecule
振子强度f的大小能反映电子跃迁能力的强弱,振子强度f越大说明跃迁能力越强,由表4可以看出,第2、8、9激发态无外电场时的振子强度f趋近于零,属于禁阻跃迁,但随着外电场强度的增大,振子强度f也有所增大,而第3、4、5激发态在无外电场时f不为零,可以发生跃迁,在特定外场下,振子强度f=0,属于禁阻跃迁,而其他激发态的振子强度在不同强度外电场作用下也发生了不同程度的变化,表明电子跃迁概率受电场影响比较复杂. 随电场强度的增加,第1、2、5、6、8激发态的激发能逐渐减小,且第1激发能减小得最快,F= 0 a.u.时的激发能将近是F= 0.03 a.u.激发能的3倍,而第3、4、7、9激发态的激发能随外电场的增加而先有微小的增大再迅速减小,总体表明外电场作用下2-氟-5-溴吡啶分子更容易被激发. 从激发波长随着外电场的变化情况可以看出,除了第3、4、7、9激发态外,其它激发态的激发波长都随电场增大而逐渐增长,特别在外电场F=0.03 a.u.时,第1激发态的激发光跃变为可见光,其它激发光仍属于紫外光.
4 结 论
本文采用密度泛函B3LYP方法和CIS/6-311++g(3df,3pd)方法对不同外电场下2-氟-5-溴吡啶分子结构和激发特征进行了研究,结果表明外场对2-氟-5-溴吡啶分子的几何结构、能级分布和激发特征都有一定的影响,结论如下:
(1)2-氟-5-溴吡啶分子的几何结构受外场的影响比较大,其中5C-9Br和3C-10F间键长受到X轴向外场影响最大,可以预见,随着外电场的继续增加,最先趋于断裂的可能是5C-9Br键和3C-10F键.
表4 2-氟-5-溴吡啶分子的激发波长与振子强度随外场的变化
Table 4 The variations of excitation wavelength and oscillator strengths in external electric fields for 2-fluoro-5-bromopyridine molecule
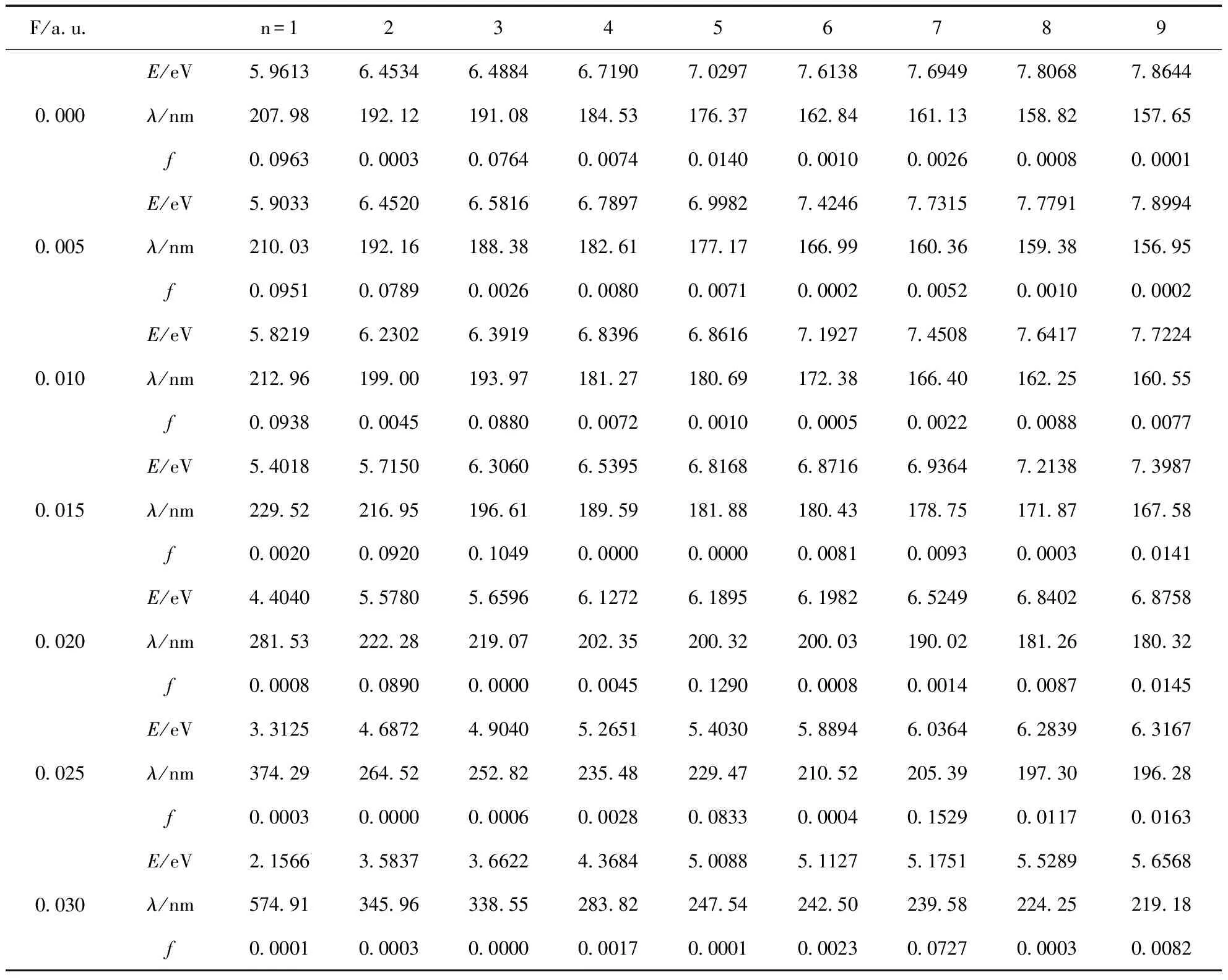
F/a u n=1234567890 000E/eV5 96136 45346 48846 71907 02977 61387 69497 80687 8644λ/nm207 98192 12191 08184 53176 37162 84161 13158 82157 65f0 09630 00030 07640 00740 01400 00100 00260 00080 00010 005E/eV5 90336 45206 58166 78976 99827 42467 73157 77917 8994λ/nm210 03192 16188 38182 61177 17166 99160 36159 38156 95f0 09510 07890 00260 00800 00710 00020 00520 00100 00020 010E/eV5 82196 23026 39196 83966 86167 19277 45087 64177 7224λ/nm212 96199 00193 97181 27180 69172 38166 40162 25160 55f0 09380 00450 08800 00720 00100 00050 00220 00880 00770 015E/eV5 40185 71506 30606 53956 81686 87166 93647 21387 3987λ/nm229 52216 95196 61189 59181 88180 43178 75171 87167 58f0 00200 09200 10490 00000 00000 00810 00930 00030 01410 020E/eV4 40405 57805 65966 12726 18956 19826 52496 84026 8758λ/nm281 53222 28219 07202 35200 32200 03190 02181 26180 32f0 00080 08900 00000 00450 12900 00080 00140 00870 01450 025E/eV3 31254 68724 90405 26515 40305 88946 03646 28396 3167λ/nm374 29264 52252 82235 48229 47210 52205 39197 30196 28f0 00030 00000 00060 00280 08330 00040 15290 01170 01630 030E/eV2 15663 58373 66224 36845 00885 11275 17515 52895 6568λ/nm574 91345 96338 55283 82247 54242 50239 58224 25219 18f0 00010 00030 00000 00170 00010 00230 07270 00030 0082
(2)2-氟-5-溴吡啶分子总能量、偶极矩和前线轨道能量也受外场较大影响. 在外电场由-0.03 a.u.变化到0.03 a.u.过程,总能量先增加再减少,偶极矩先减小后增加,轨道能隙则先急剧增大,再缓慢减小,在急剧减小.
(3)无外电场的情况下,2-氟-5-溴吡啶分子的第2、8、9激发态趋近于禁阻跃迁,其他激发态都能够激发;随着外电场强度的增大,第2、8、9激发态变得能够激发,而第3、4、5激发态在一定外电场下振子强度f=0,又属于禁阻跃迁. 另外,外电场对2-氟-5-溴吡啶分子的激发波长也产生了很大的影响.
[1] Guo Y, Xie D Q, Xue Y,etal. Density functional theory studies on vibrational spectra of 2-fluoro-5-bromopyridine[J].ActaChimicaSinica, 2002,(4): 660 (in Chinese) [郭勇, 谢代前, 薛英, 等. 2-氟-5-溴吡啶分子振动光谱的密度泛函理论研究[J]. 化学学报,2002,(4): 660]
[2] Chen H Q, Wan L B, Chen X Q. Progress in synthesis of 2-fluoropyridine[J].SpecialityPetrochemicals, 2007,(6): 76 (in Chinese) [陈海群, 万利兵, 陈小强. 2-氟吡啶合成的研究进展[J]. 精细石油化工, 2007 (6): 76]
[3] Wang Y, Chen X, Qi Y,etal. Synthesis of 2-alkyloxy-5-bromopyridine[J].JournalofTianjinNormalUniversity(NaturalScienceEdition), 2006,(1): 14 (in Chinese) [王越, 陈煦, 齐艳, 等. 2-烷氧基-5-溴吡啶的合成[J]. 天津师范大学学报(自然科学版), 2006,(1): 14]
[4] Zhou F L, Peng X J, Chen H W,etal. Study on synthesis and behavior of adsorbing gold of 5-brom pyriclyiazo-hacid[J].JournalofEastChinaGeologicalInstitute, 2000,(4): 282 (in Chinese) [周发连, 彭雪娇, 陈焕文, 等. 5-溴吡啶偶氮-H螯合树脂合成及吸附金性能研究[J]. 华东地质学院学报, 2000, (4): 282]
[5] Fang Y Q, Sun D X. Synthesis of 2-amino-5-bromopyridine [J].SpecialityPetrochemicals, 2010, (7): 4 (in Chinese) [方永勤, 孙德鑫. 2-氨基-5-溴吡啶的合成[J]. 精细石油化工, 2010, (7): 4]
[6] Chen X J, Ma M Z, Luo S Z,etal. Exploring the effects of an external electric field on the acetylene[J].J.At.Mol.Phys., 2004, 21(1): 19 (in Chinese) [陈晓军, 马美仲, 罗顺忠, 等. 外电场作用下乙炔分子的激发态研究[J]. 原子与分子物理学报, 2004, 21(1): 19]
[7] Wu X K, Liang D M, Jing T. Study on the properties of L1OH molecule under the external electric field[J].J.At.Mol.Phys., 2014, 31(2): 187 (in Chinese) [吴学科, 梁冬梅, 荆涛. 外电场作用下L1OH分子的特性研究[J]. 原子与分子物理学报, 2014, 31(2): 187]
[8] Xu G L, Zhu Z H, Ma M Z,etal. Study on the effect of external electric field excitation on methane[J].ActaPhys.Sin., 2005, 54(7): 3087 (in Chinese) [徐国亮, 朱正和, 马美仲, 等. 甲烷在外场作用下的光激发特性研究[J]. 物理学报, 2005, 54 (7): 3087]
[9] Grozema F C, Telesca R, Joukman H T. Excited state polarizabilities of conjugated molecules calculated using time dependent density functional theory[J].Chem.Phys., 2001, 115: 10014.
[10] Kjeellberg P, Zhi H, Tunu P. Bacteriochlorophyll in electric field[J].J.Phys.Chem. B, 2003, 107: 13737.
[11] Huang D H, Wang F H, Min J,etal. Study on structure characteristics of MgO molecule under external electric field[J].ActaPhys.Sin., 2009, 58(5): 3052 (in Chinese) [黄多辉, 王藩侯, 闵军, 等. 外电场作用下MgO分子的特性研究[J]. 物理学报, 2009, 58 (5): 3052]
[12] Ling Z G, Tang Y L, Li T,etal. Molecular structure and properties of zirconiumdioxide under the external electric field[J].ActaPhys.Sin., 2014, 63(2): 023102 (in Chinese) [凌智钢, 唐延林, 李涛, 等. 外电场下二氧化锆的分子结构及其特性[J]. 物理学报, 2014, 63(2): 023102]
Influence of external electric field on the molecular structure and electronic spectrum of 2-fluoro-5-bromopyridine molecule
WU Xue-Ke1, ZHANG Song1, LIANG Dong-Mei1,2
(1.School of Physics and Electronic Engineering, Kaili University, Kaili 556011,China; 2. School of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China)
The B3LYP method of density functional theory (DFT) at 6-311++g(3df,3pd) level is used to calculate the geometrical parameters, dipole moments and total energies of the ground state of 2-fluoro 5-bromopyridine molecule under different external electric fields in this paper. On the basis of this, the single-excitation configuration interaction (CIS) is used to study the influences of external electric field on the excited wavelengths and oscillators of the first nine excited states. The results show that the bond lengths (5C-9Br, 3C-10F) will be greatest impact by X axial electric field, and with the continue increase of electric field, they may be the first to break. When the external electric field F=0.005 a.u., the total energy is maximum, and the dipole moment is minimized. The LUMO energy levels are greatly influenced by external electric field, but the HOMO energy levels are less affected by the external electric field. The excited wavelength and oscillator are influenced by electric field, but their changes with the electric field are relatively complex.
2-fluorio-5-bromopyridine molecule; Excited state; External electric fields
贵州省科学技术基金(黔科合J字[2013]2262号);凯里学院规划课题(Z1409,Z1405);贵州省普通高等学校创新人才团队(黔教合人才团队字[2012]06号)
吴学科(1979—),男,贵州黎平人,硕士,副教授,主要从事原子分子物理等研究.E-mail: wuxueke@sina.com
103969/j.issn.1000-0364.2015.08.009
0561; 0433
A
1000-0364(2015)08-0579-07
投稿日期:2014-08-07
