Cu-Co双金属团簇结构演化及其性质的分子动力学模拟
2015-03-23孙凌涛石东平
孙凌涛,石东平
(重庆文理学院新材料技术研究院,重庆 402160)
Cu-Co双金属团簇结构演化及其性质的分子动力学模拟
孙凌涛,石东平
(重庆文理学院新材料技术研究院,重庆 402160)
采用分子动力学结合嵌入原子方法对比研究了Co分布于Cu-Co团簇不同层的结构和性质.研究表明:Co原子分层掺杂可对团簇的结构转变点和熔点进行诱导控制;分层掺杂的Cu-Co团簇第一相变是一种扩散度较小的由立方八面体转变为二十面体的相变;Co原子易于向低能态团簇的亚表层(111)面偏析,从而诱导团簇结构紊乱,造成其熔点差异.
团簇; 结构; 偏析; 分子动力学
1 引 言
介于原子、分子与块体之间的一种物质新层次,团簇因较大的表体比引起的表面效应,会表现出许多特殊的物理化学性质[1].由两种金属原子聚集而成的双金属团簇,可以通过异质原子成分、尺寸和排列方式对团簇的物理、化学和电磁性能进行调节[2,3].在双金属纳米团簇展现的众多特殊功能中,热力学行为表现出许多奇特性质,如升温过程中团簇表现出尺寸效应[4]和分层预融[5]等.这使得双金属团簇在传感、纳米催化和微电子等新材料领域中具有广阔应用前景,相关理论[6]、实验[7]研究成为近年热点.
对于双金属团簇,为了释放内应力,小原子比大原子有更强烈的向核分布趋势,这种效应在二十面体团簇中较为强烈,而在FCC和十面体团簇中较弱.另外,为了降低粒子的表面能,双金属团簇中表面能较小的原子易于向表面偏析[8].因此任意两种金属原子由于原子尺寸和表面能的差异会使团簇在热力学过程中出现偏析现象,从而诱发团簇结构形貌、表面组分及性质发生改变[9].最近的理论和实验研究发现,原子偏析会使一些双金属体系易于形成表面能小的原子包裹表能较大原子的核壳结构[10,11].例如,通过温控,Ag原子偏析会诱导Co-Ag团簇形成具有近似hcp核的核壳结构[12],也可以形成具有Ag-Co-Ag的洋葱型结构[13].通过在Au团簇表面包裹Ag原子,可以大幅降低Ag-Au团簇马氏体转变温度[14].以不同比例掺杂的Cu-Co双金属团簇以一定冷却速率降温后,由于原子偏析会诱导其形成特定的可控结构[15,16].以Co原子为基体的Co-Rh团簇,部分Co原子偏析到表面,另外一部分仍占据于核心层,这将诱导团簇形成洋葱型结构[17].目前原子偏析作为一个研究侧入点,主要侧重于对双金属团簇降温过程中团簇结构形貌的研究,但对升温过程中原子偏析诱导的团簇结构、形貌、相变点变化的作用细节和机理还不够深入.另外,目前的研究多采用表面能小的原子随机掺杂或按层分布于表面能较大的基体团簇,来考察表面能小的原子向双金属团簇表面偏析对团簇的结构和性质的影响,对表面能大的原子随机或分层掺杂于表面能小的团簇中研究还较少.
针对目前研究中存在的问题,本文以表面能相差较大的Cu(1592 mJ/m2)和Co(2197 mJ/m2)[18]原子,构建了Co原子分布于基体Cu团簇不同层的双金属团簇.然后,采用分子动力学模拟方法,并结合势能-温度曲线、团簇快照图、对分布函数及Honeycutt-Andersen(HA)指数[19]等不同表征分析手段,对升温过程中Co分层掺杂Cu-Co团簇的结构及性质进行了研究.
2 计算方法
团簇模型采用五阶Gear预报-校正算法积分牛顿方程,用标度方法控制系统的温度.模拟系统使用正则系综(NVT)且无外力的周期性边界条件.由于截断半径选取的差异,模拟纯金属团簇的势函数不适合模拟异质二元合金团簇,本文使用由Zhou等[20,21]提出的一种合金原子间的相互作用—镶嵌原子势(EAM),它采用了和嵌入原子法相同的分析形势,可用来研究16种任意组合金的相互作用.团簇热力学模拟过程将团簇从300 K连续升温至1400 K,升温速率0.6 K/ps,模拟时间步长0.5 fs.含有309个原子初始团簇是从10a0×10a0×10a0(晶格常数a0=0.3615)的FCC块体中截取,然后分别用Co原子替换立方八面体Cu团簇亚表层及次表层的Cu原子所得.Co分布于Cu团簇次亚表层(简称:次表Co团簇)和亚表层(简称:亚表Co团簇)的初始团簇的结构形貌及其Cu和Co原子的径向密度函数图1中(a)、(b)所示.
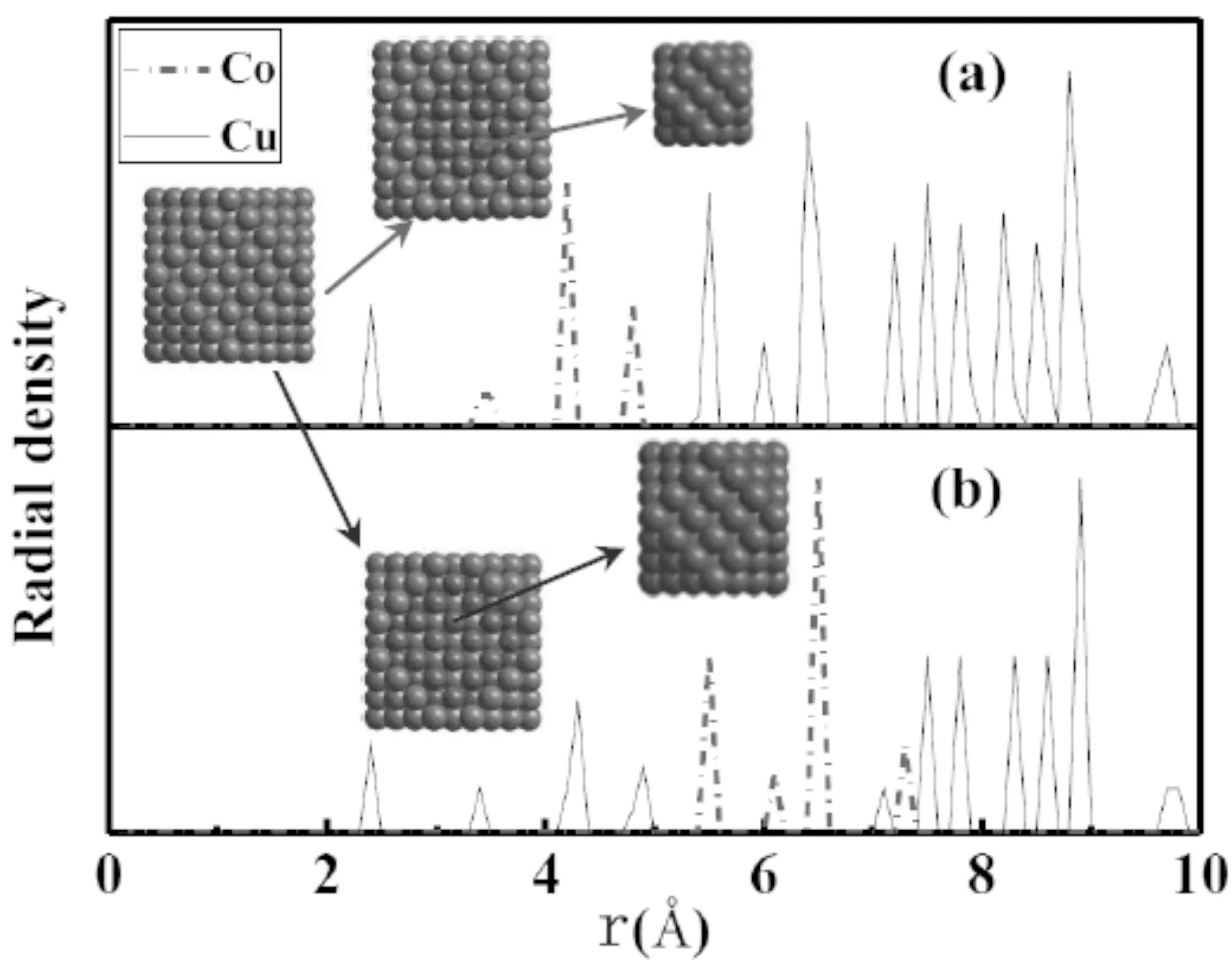
图1 团簇中Cu和Co原子的径向分布密度及各层的表面形貌Fig.1 Atomic radius distributions of the clusters and their corresponding initial morphologies
3 结果与讨论
首先给出了升温过程中纯Cu及其两种掺杂团簇温度-能量曲线,如图2所示.可以看出,随着温度的升高,三种组分的团簇存在相同的变化趋势,都会在低温时出现势能向下跳跃点,在高温时出现势能向上跳跃点,在跳跃点前后团簇的能量与温度呈近似线性关系.所不同的是纯Cu团簇和次表Co团簇的第一个势能拐点出现在300-400 K之间,而亚表Co团簇的第一势能拐点出现在400-500 K之间;纯Cu团簇、次表Co团簇及亚表Co团簇的第二个拐点分别出现在800-900 K、900-1000 K、1000-1100 K之间.从图2纵向来看,纯Cu团簇的平均势能整体高于亚表Co及次表Co团簇,亚表Co团簇能量相对较低.

图2 升温过程中团簇原子的温度-能量曲线Fig.2 Variation of energy with temperature for clusters
图2中势能拐点位置及其能量纵向差异说明Co原子分层掺杂会直接影响到团簇的结构转变、熔点及能量.用势能-温度曲线来确定团簇的相变点非常直观,但是由于势能拐点的跳跃区间覆盖了一个范围,无法精准锁定某一温度值.基于此,还需用温度-热熔曲线来精确化结构相变点,如图3所示.可以看出三种组分团簇的相变点位置存在较大的差异,亚表Co团簇、次亚Co团簇及纯Cu团簇第一相变点分别为428 K、370 K和377 K,相应的第二相变点分变为1008 K、934 K及853 K.

图3 团簇的热容-温度曲线Fig.3 Temperature dependence of the heating capacity for clusters
为了考察团簇相变点的团簇结构变化,图4给出了亚表Co团簇分别在第一相变点前后的对分布函数(图中a、b所示)和次表Co团簇在第一相变点前后的对分布函数(图中c、d所示).可以看出,亚表Co团簇转变前后对分布函数峰位、峰强度都有着明显的差异,相对于转变前来说,转变后的第三峰明显减小,从第四峰开始,转变后的峰位开始错位并逐渐消失,说明转变前后结构发生了明显变化,结合快照图考察,亚表Co团簇由FCC结构转变为具有明显五重对称轴的二十面体结构.对于次表Co团簇,其转变前后的对分布函数分别与亚表Co团簇转变前后的对分布函数有着相同的趋势,他们的峰位和峰强基本一致,这说明次表Co团簇转变前后也分别是FCC结构和二十面体结构,对应的结构快照图验证了这一点.总的来看,亚表Co团簇和次表Co团簇,尽管都发生了从FCC到二十面体的结构转变,但其结构中Co原子所处层位基本没变化,说明这种结构转变是一种扩散度较小的相变.另外,由于二十面体较立方八面体结构更稳定,所以团簇在从立方八面体向二十面体结构转变的过程中势能会降低,从而说明了图2中一级相变点势能向下跳跃的原因.

图4 亚表Co团簇和次表Co团簇在第一相变点前后的对分布函数Fig.4 Pair correlation function of subsurface cluster and 3rd-shell cluster at the temperature before and after the first phase transition point
键对分析方法已被广泛用于监测液体、玻璃态及非晶结构,这种技术主要采用原子与近邻原子的关系(HA指数)来考察团簇升温过程中整体的结构变化,图5、6分别给出了亚表Co团簇和次表Co团簇的HA指数随温度变化的关系曲线,其中1421键表征面心立方结构,1422键表征体心立方结构,1551键表征二十面体结构,1541健表征缺陷及无序结构.图5中在300 K时1551键值为0,而1421键值为0.73.在370 K左右,亚表Co团簇的HA指数有明显的变化,其中1421键值变为0.29,团簇已不再具有FCC结构,相应1551键值由0变为0.03,此时团簇已由FCC结构转变为二十面体结构,这和图4结论一致.从300 K到熔点934 K之后,团簇的1551、1541、1421及1422键值相差幅度由大变小,最后基本接近,这是由于从低温到高温团簇由固体变为液体熔化所造成的.图6中亚表Co团簇HA指数在升温区间内也存在明显的两次跳跃,但其变化区间在400-500 K及1000-1100 K之间.在一级相变点,亚表Co团簇在转变前1421键值为0.67,转变后1421明显降低,此时1551键值为0.03团簇此时已转变为二十面体.在熔点后,团簇各HA指数大小接近,此时团簇已熔化.

图5 亚表Co团簇HA指数随温度的变化Fig.5 HA index as a function of temperature for the subsurface cluster
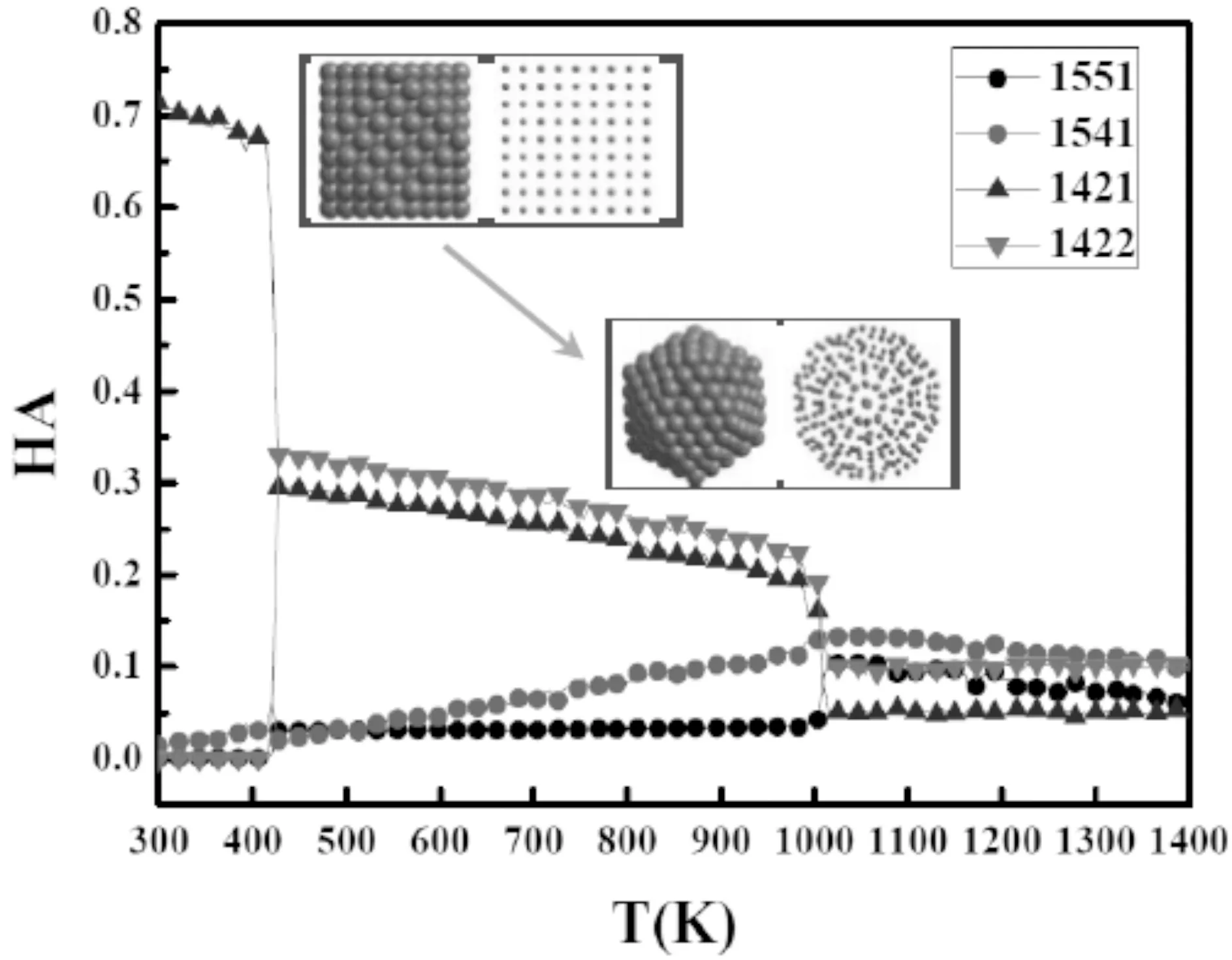
图6 次表Co团簇HA指数随温度的变化Fig.6 HA index as a function of temperature for the 3rd-shell cluster
为了研究团簇分层掺杂产生的结构转变点及熔点的差异,对Co分布于团簇中不同位置点的能量进行了分析.主要的技术手段是用一个Co原子替换纯Cu二十面体团簇中每一层不同位置(棱边、顶点、(111)面)的Cu原子,然后将替换后的团簇在0K下弛豫1ns得到团簇每个原子的平均能量(见图7).从图中可以发现,Co原子分布于亚表层(111)面时能量最低,并且其处于表层时的原子能量远高于其它各层.Co原子位于亚表层时,在棱边的能量最高,其次是顶点,最低的是(111)面;Co原子位于次表层时,Co在顶点和棱边的能量相同且略低于(111)面.Co原子这种不同分布,会造成三个结果,一是Co原子分布在Cu团簇的亚表层(111)面的能量低于Co分布在次表层(111)面,这会导致要使亚表Co团簇熔化需要更高的温度,这与图3所得结果取得一致.二是随着温度的升高,亚表及次表Co团簇中的Co原子位置在各层顶点、棱边和(111)面变化会导致团簇能量不断变化,这与图2曲线取得一致.三是随着温度的升高,次亚表层Co团簇中的Co原子易于偏析到团簇的亚表层,而亚表层Co团簇中的Co原子位置易于从顶点和棱边向(111)面变化.
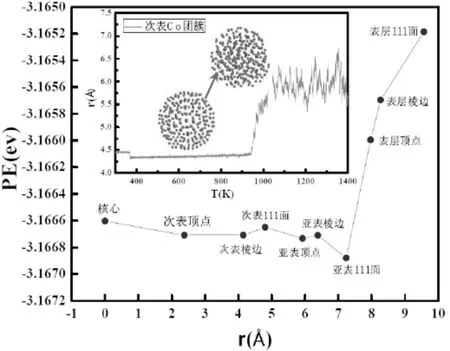
图7 团簇在0 K弛豫1 ns后原子的平均能量及此表Co团簇升温过程中的原子能量和Cu原子与团簇质心的间距Fig.7 The average atom energy of clusters at 0 K and relaxation after 1 ns,the atomic energy in the process of Co clusters on the second floor heating and the distance from mass center of Cu atom to cluster
为了探究团簇熔点差异的原因,图7附图还给出了Co原子与团簇质心的平均距离随温度变化函数.由于原子偏析会使团簇局部晶格错配,且处在亚表层的Co原子发生偏析时其位置很难精确定位,于是附图中只给出了次表Co团簇中Co原子温度-离质心平均距离关系,其中Co原子与质心的平均距离越大说明偏析过程越明显.可以看出,团簇在300-400 K之间有一个向下的跳跃,但这种转变量较小,说明此时Co原子位置有一个较小的变化,这和图4结论“Co原子所处层位基本没变化,说明这种结构转变是一种扩散度较小的相变”取得一致.在400-900 K之间团簇中Co的原子的相对位置基本没变,团簇的形貌基本保持二十面体结构.在900-1000 K之间,随着温度升高,Co原子离质心的距离有一个突然的激增,表明大量Co原子开始从次亚表层向亚表层偏析,且在934 K团簇由二十体结构转变为无序结构,表面团簇已经熔化,这说明Co原子的偏析伴随于团簇熔化过程中,是造成团簇熔点差异的主要诱因.
4 结 论
本文采用分子动力学结合嵌入原子方法,对比研究了升温过程中亚表及次表Co团簇的结构和性质.研究结果表明:分层掺杂会使纯Cu团簇、次表Co团簇及亚表Co团簇的结构转变点和熔点有差别,说明分层掺杂可以对团簇的结构转变点和熔点进行诱导,这为制备可控熔点的双金属团簇提供一条有效途径;分层掺杂的Cu-Co团簇第一相变是一种扩散度较小的由立方八面体转变为二十面体结构的相变;由于分布在团簇各层不同顶点、棱边、(111)面上的Co原子能量的不同,会使Co原子易于向低能态的团簇亚表层(111)面偏析,从而诱导团簇结构紊乱,造成其熔点差异.
[1] Wang G H.Clusterphysics[M].Shanghai: Shanghai Scientific & Technical Publisher, 2003(in Chinese)[王广厚. 团簇物理学[M]. 上海: 上海科学技术出版社,2003]
[2] Ferrando R,Jellinek J,Johnston R L. Nanoalloys: from theory to applications of alloy clusters and nanoparticles[J].Chem.Rev., 2008,108(3): 845.
[3] Mariscal M M,Dassie S A,Leiva E P M. Collision as a way of forming bimetallic nanoclusters of various structures and chemical compositions[J].J.Chem.Phys., 2005,123(3): 184505.
[4] Kart H H,Yildirim H,Ozdemir K S,etal.Physical properties of Cu nanoparticles:A molecular dynamics study[J].Mater.Chem.Phys., 2014,147(1-2): 204.
[5] Lewis L J,Jensen P,Barrat J L. Melting,freezing,and coalescence of gold nanoclusters[J].Phys.Rev. B,1997,56(4): 2248.
[6] Baletto F,Ferrando R. Structural properties of nanoclusters: Energetic,thermodynamic,and kinetic effects[J].Rev.Mod.Phys., 2005,77(1): 371.
[7] Schmidt M,Kusche R,Issendorff B V,etal. Irregular variations in the melting point of size-selected atomic clusters[J].Nature,1998,393(6682): 238.
[8] Yang J Y,Hu W Y,Wu Y R,etal. Substrate dependence of growth configurations for Co-Cu bimetallic clusters[J].Cryst.GrowthDes., 2012,12(6): 2978.
[9] Chushak Y G,Bartell L S. Freezing of Ni-Al bimetallic nanoclusters in computer simulations[J].J.Phys.Chem. B,2003,107(16): 3747.
[10] Song P X,Wen D S. Molecular dynamics simulation of a Core-Shell structured metallic nanoparticle[J].J.Phys.Chem. C,2010,114(19): 8688.
[11] Ferrer D,Torres-Castro A,Gao X,etal. Three-layer core/shell structure in Au-Pd bimetallic nanoparticles[J].Nano.Lett., 2007,7(6): 1701.
[12] Dorfbauer F,Schrefl T,Kirschner M,etal. Nanostructure calculation of CoAg core-shell clusters[J].J.Appl.Phys., 2006,99(8): 08G706.
[13] Wang Q,Li G J,Li D G,etal. Evolution of three-shell onion-like and core-shell structures in(AgCo)201 bimetallic clusters[J].Chin.Phys. B,2009,18(5): 1843.
[14] Chen F Y,Johnston R L.Martensitic transformation in Ag-Au bimetallic core-shell nanoalloys[J].Appl.Phys.Lett., 2008,92(2): 023112.
[15] Zhang Y J,Li Y Q,Xiao X Y,etal. Effect of Cu atomic segregation on the frozen structures of Co-Cu bimetallic clusters[J].NANO:BriefReportsandReviews,2012,7(6): 1250047.
[16] Li G J,Wang K,Wang Q,etal. Formation of icosahedral and hcp structures in bimetallic Co-Cu clusters during the freezing processes[J].Mater.Lett., 2012,88: 126.
[17] Fromen M C, Morillo J,Casanove M J,etal. Structure and chemical order in Co-Rh nanoparticles[J].Epl-Europhys.Lett., 2006,73(1): 885.
[18] Nanda K K,Sahu S N,Behera S N. Liquid-drop model for the size-dependent melting of low-dimensional systems[J].Phys.Rev. A,2002,66(1): 013208.
[19] Honeycutt J D,Andersen H C. Molecular dynamics study of melting and freezing of small Lennard-Jones clusters[J].J.Phys.Chem., 1987,91(19): 4950.
[20] Zhou X W,Wadley H N G,Johnson R A,etal. Atomic scale structure of sputtered metal multilayers[J].Acta.Mater., 2001,49(19): 4005.
[21] Wadley H N G,Zhou X,Johnson R A,etal. Mechanisms,models and methods of vapor deposition[J].Prog.Mat.Sci., 2001,46(5): 329.
Molecular dynamics simulation on the structure evolution of bimetallic Cu-Co clusters and their properties
SUN Ling-Tao,SHI Dong-Ping
(Research Institute for New Materials Technology,Chongqing University of Arts and Sciences,Chongqing 402160,China)
A comparison study on the structures and properties of the Co atom distribution in different layers of Cu-Co clusters was performed by the molecular dynamics combining with atom-embedded method.Research results showed that the layer-doping of Co atom can induce and control the structural transformation point and melting point of clusters.The first phase transition of layer-doped Cu-Co clusters is a smaller diffusion degree of phase transformation from cubic octahedron into icosahedron.Additionally,Co atom is easy to have the surface segregation tendency to the subsurface(111)of the low-energy state of clusters,further resulting in the structural disorder of the cluster and the difference of the melting point.
Clustes; Structure; Segregation; Molecular dynamics
2014-12-09
重庆文理学院校级科研项目(Y2013CJ26); 重庆市高校微纳米材料工程与技术重点实验室度开放课题(KFJJ1404)
孙凌涛(1987—), 男, 宁夏中卫人, 硕士, 助教, 主要研究方向为材料物理与化学.E-mail: cquptslt@163.com
103969/j.issn.1000-0364.2015.08.010
O561
A
1000-0364(2015)08-0586-05
