不同磷脂肽段诱导的实验性自身免疫性脑脊髓炎模型病理特征多样性的研究①
2015-03-18郑梅梅王拥军张星虎首都医科大学附属北京天坛医院神经内科北京100050
郑梅梅 王拥军 张星虎 (首都医科大学附属北京天坛医院神经内科,北京100050)
多发性硬化(Multiple sclerosis,MS)是最常见的中枢神经系统(Central nervous system,CNS)脱髓鞘疾病,临床表现多种多样[1],包括肢体无力、感觉障碍、视力下降、共济失调、发作性症状等,也可表现为疲劳、抑郁等非特异性症状,任何两位MS 患者的临床表现都不尽相同。MS 的临床特点之一是时间多发性,85%~90%的MS 患者常常经历病情的反复发作和缓解;另一特点是病灶在空间上具有多发性,视神经、大脑、小脑、脑干和脊髓均可受累[2]。MS的影像学表现各异,位于侧脑室周围白质的病灶比较常见,其长轴常与侧脑室垂直,呈圆形、卵圆形。约2/3 患者可出现皮质下白质病变,有时病灶也可表现为黑洞、同心圆,当胼胝体病变时可呈现“虫蚀状”[3]。MS 病理特点为髓鞘脱失、炎性细胞浸润、星形胶质细胞增生,而轴索相对保留[4]。Bruck等[1]认为MS 免疫病理学特征具有人群异质性,即不同的患者具有不同免疫病理学特征,且根据病灶的免疫病理学参数的异质性将病灶分为4 种组织病理学类型。以此分型为基础,研究认为同一患者的病灶只表现为一种病理类型,即病灶的免疫病理学特征在某一患者中具有同质性[1]。
但MS 患者临床表现差异、病理学人群异质性的原因尚不清楚。常采用磷脂抗原免疫易感动物建立实验性自身免疫性脑脊髓炎(Experimental autoimmune encephalomyelitis,EAE)动物模型进行MS相关研究。常用的致敏抗原是髓鞘碱性蛋白(Myelin basic protein,MBP)和髓鞘少突胶质细胞糖蛋白(Myelin oligodendrocyte glycoprotein,MOG),MBP 是髓鞘成分中抗原性最强的蛋白质,MOG 既可诱导自身抗体反应,也可激活致脑炎性T 细胞[5]。
Berger 等[6]发现不同磷脂肽段特异性T 细胞诱导的Lewis 大鼠EAE 模型具有抗原特异性炎性细胞浸润分布,而且不同的磷脂肽段是致脑炎自身反应性T 细胞作用的靶点,神经组织实质炎性细胞浸润的程度和巨噬细胞活化的程度也与免疫抗原有关[7]。Abromson-Leeman 发现[8]MBP59-76 特异性T细胞免疫大鼠只可产生上升性脊髓麻痹,而MBP151-168 特异性T 细胞可以诱导周围神经系统的炎症和脱髓鞘,提示T 细胞的抗原表位特异性与病灶分布可能具有关联。MBP68-86 肽段诱导Lewis大鼠后,在脊髓发现T 细胞的浸润和小胶质细胞的激活[9]。MOG35-55 可诱导侧脑室旁、丘脑、脑干、小脑和上段颈髓的病灶[10]。
CNS 局部黏附分子和趋化因子的不同可能与这些部位是否出现病灶有关[11]。CCL-7 是一种趋化因子,可趋化单核细胞和淋巴细胞,在多种CNS炎性疾病中其mRNA 表达均上调。血管内皮黏附分子-1(Vascular cell adhesion molecule-1,VCAM-1)是一种重要的黏附分子,可帮助细胞穿越血脑屏障,并使其在脑实质中驻留。
虽然以上研究也使用不同磷脂肽段免疫Lewis大鼠进行相关研究,但是存在以下局限性:(1)易感动物的遗传学差异、体重、性别影响病理学表现,但以上研究所使用的Lewis 大鼠并不来源于同一克隆,且体重、性别均各不相同;(2)上述研究仅观察部分病理学表现,未全面观察炎性细胞浸润、脱髓鞘和轴索受累等病理学特点。综上,本课题组拟采用3 种不同磷脂肽段免疫具有相同遗传背景的Lewis大鼠,观察动物模型的神经功能评分、炎性细胞浸润的部位和程度、脱髓鞘分级和有无轴索受累,同时探索病灶局部黏附分子和趋化因子基因的表达情况。
1 材料与方法
1.1 实验动物 Lewis 大鼠(雌性,6~8 周,160~180 g,SPF 级)购自北京维通利华实验动物技术有限公司。饲养于首都医科大学实验动物中心实验室,环境设有恒温、恒湿和除菌换气系统,具有严格的微生物控制系统。大鼠3 只一笼,光照周期为12/12 h。本研究严格遵循《实验动物管理条例》,经首都医科大学伦理委员会批准(批准号:2012-X-87)。所有手术操作均采用水合氯醛10%麻醉(1 ml/100 g)。
1.2 主要试剂 豚鼠MBP68-86:序列YGSLPQKSQRSQDENPV。大鼠MBP82-99:序列DENPVVHFFKNIVTPRTP。大鼠MOG35-55:序列MEVGWYRSPFSRVVHLYRNGK。以上肽段纯度>99%,购自上海吉尔生化有限公司。不完全弗氏佐剂(Freund's incomplete adjuvant,IFA)购自美国Sigma 公司,抗NF-200 抗体(Neurofilament-200,抗神经丝蛋白-200抗体)购自Abcam 公司,Trizol 试剂购自美国Invitrogen 公司,反转录试剂盒和RT-PCR 试剂盒购自大连TaKaRa 公司。
1.3 实验性自身免疫性脑脊髓炎大鼠模型建立 IFA 加入人结核分枝杆菌(H37Ra)制备成含有4 mg/ml H37Ra 的弗氏完全佐剂(Freund's complete adjuvant,CFA)。CFA 与等体积的含有1 mg/ml 的MBP68-86、1 mg/ml 的 MBP82-99、2 mg/ml 的MOG35-55 的磷酸盐缓冲液(Phosphate buffered saline,PBS,pH7.4)均匀混合制备免疫乳剂。将大鼠随机分为5 组,分别为正常对照组、佐剂对照组、MBP68-86 组、MBP82-99 组、MOG35-55 组、每组20只。向大鼠两侧尾根皮下分别注射乳剂100 μl,总计每只大鼠注射乳剂200 μl。正常组大鼠不接受乳剂免疫。佐剂对照组接受200 μl 的CFA 与PBS 的混合乳剂。MBP68-86 组、MBP82-99 组、MOG35-55组大鼠分别接受100 μg MBP68-86 组、100 μg MBP82-99 组、200 μg MOG35-55。
1.4 EAE 大鼠神经功能评分和体重监测 免疫后每组各取6 只大鼠每天进行神经功能评分,并测量体重。神经功能评分采用以下方法:0 分:无体征;1分:尾部无力;2 分:单个或双后肢不完全性瘫痪;3分:单个或双后肢完全性瘫痪;4 分:伴有前肢无力或瘫痪;5 分:濒死状态或死亡。
1.5 组织学观察
1.5.1 制备石蜡切片 免疫后第12~15 天(Day post immunization,dpi),每组取8 只大鼠经心脏灌注生理盐水和4%多聚甲醛。分别取出大脑、脑干、小脑和脊髓,置入4℃PFA 中放置4 h。制备石蜡组织块和组织切片。
1.5.2 HE 染色观察炎性细胞浸润 每只大鼠每个部位随机选10 张切片进行HE 染色,于×400 光学显微镜下观察每张切片炎性细胞浸润灶的数目。一个炎性细胞浸润灶定义为至少20 个炎性细胞聚集在一起[12]。采用Photoshop 软件计算每张组织切片的面积,并计算每平方厘米组织炎性细胞浸润灶的数目。
1.5.3 快蓝(Luxol Fast Blue,LFB)染色观察髓鞘每个部位另选10 张切片行LFB 染色。显微镜下观察有无髓鞘脱失。脊髓脱髓鞘的严重程度按照评分标准进行[12]。
1.5.4 免疫组化染色,观察轴索受累情况 石蜡切片采用3% H2O2阻断内源性过氧化物酶,枸橼酸修复液抗原修复,滴加一抗抗NF-200 抗体(1∶100 稀释),4℃过夜,切片不加一抗作为阴性对照。滴加二抗,DAB 显色、苏木素复染和分化,返蓝,脱水、透明和封片;镜下观察轴索受累情况,重点观察是否存在轴索肿胀、变细、断裂、卵形体形成[13]。
1.6 实时定量聚合酶链反应(RT-PCR) 采用RT-PCR 方法定量测定CCL-7、VCAM-1 和NEFM的mRNA 表达情况。免疫后第10 天,每组取6 只大鼠拉颈处死,迅速取出各组大鼠大脑、小脑、脑干和脊髓,快速放入液氮中灭活RNA 酶。采用Trizol 试剂提取各部位全基因组RNA。去除DNA,反转录成cDNA。实时定量PCR:CCL-7 前向引物序列(GGGACCAATTCATCCACTTGC),反向引物序列(TCAGCACAGACTTCCATGCC)。VCAM-1 前向引物序列(GGAAATGCCACCCTCACCTT),反向引物序列(CACCTGAGATCCAGGGGAGA)。NEFM前向引物序列(TCTGTACACACCCGAA-CAGC),反向引物序列(CTGTGAGGGCGTCTTCA-CATT)。94℃反应5 min;变性和扩增和延伸,94℃反应30 s,58℃反应30 s,72℃反应40 s,共40 个循环;72℃反应7 min。采用ΔΔcycle threshold (ΔΔCt)方法进行定量分析[14]。基因拷贝数经管家基因3-磷酸甘油醛脱氢酶(Glyceraldehyde-3-phosphate dehydrogenase,GAPDH)的拷贝数校正。计算2ΔΔCt值,即为实验组的目的基因表达量是对照组表达量的百分数,大于200%为表达上调,小于50%为表达下调。
1.7 统计学分析 使用SPSS16.0 进行分析。ANOVA 和Student's T-test 用于组间差异比较。数据以±s 表示。组间神经功能评分比较采用非参数Mann-Whitney U 检验。采用Pearson 相关分析或者偏相关分析方法进行相关分析。以P <0.05 为差异具有统计学意义。
2 结果
2.1 EAE Lewis 大鼠神经功能评分变化规律 MBP82-99 组和MBP68-86 组大鼠发病率为100%,MOG35-55 组和对照组大鼠均未表现出EAE 的临床体征。MBP68-86 组大鼠的神经功能评分较MBP82-99 组大鼠高(P <0.05)。各组大鼠神经功能评分变化趋势见图1。
2.2 组织学表现
2.2.1 神经组织炎性细胞浸润情况 对照组大鼠各神经组织均未见到炎性细胞浸润。MBP68-86 组、MBP82-99 组两组大鼠的大脑、脑干、小脑和脊髓组织均发现炎性细胞浸润,以小血管周围和软脊膜下为主。MOG35-55 组大鼠仅有脊髓组织存在炎性细胞浸润。HE 代表性图片见图2A。各组大鼠不同部位神经组织炎性细胞浸润灶数目/cm2见图2B。各组大鼠脊髓组织炎性细胞浸润数目均高于其余部位(P <0.01),且MBP68-86 组和MBP82-99 组的脊髓组织炎性细胞浸润数目高于MOG35-55 组大鼠(P<0.01)。
本研究还分析了MBP82-99 组大鼠神经功能评分与脊髓实质炎性细胞浸润数目/cm2之间的关系,发现神经功能评分与脊髓实质炎性细胞浸润数目呈正相关(r=0.494)。
2.2.2 神经组织脱髓鞘和轴索受累观察 各组大鼠神经组织LFB 染色均未见任何脱髓鞘改变,见图3。本研究各组大鼠也未见轴索受累。
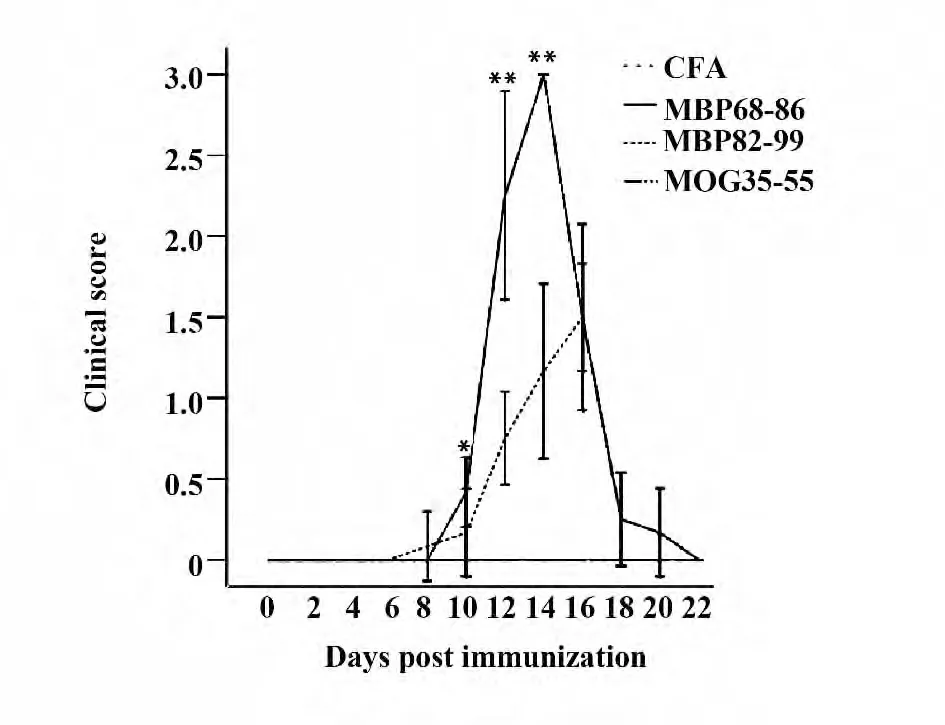
图1 免疫后各组大鼠的神经功能评分变化Fig.1 Neurological score of EAE rats induced by MBP68-86,MBP82-99,MOG35-55 and CFA only
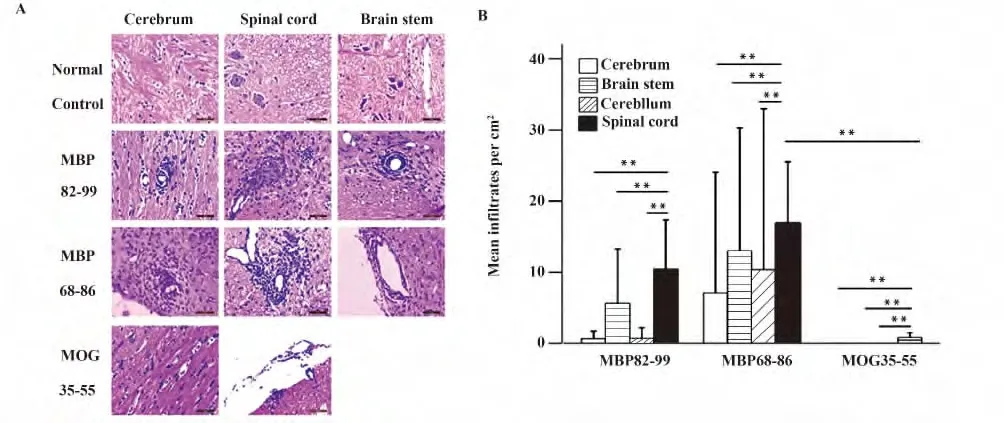
图2 大鼠神经组织HE 染色的代表性图片,不同部位神经组织炎性细胞浸润灶数目/cm2(×400)Fig.2 Inflammatory infiltrates in CNS of rats at 12 -15th day post immunization(×400)
图3 MBP68-86 诱导的EAE 大鼠脊髓组织LFB 髓鞘染色图片,未见脱髓鞘现象(×400)Fig.3 LFB staining of spinal cord of MBP68-86 induced EAE didn't showed demyelination(×400)
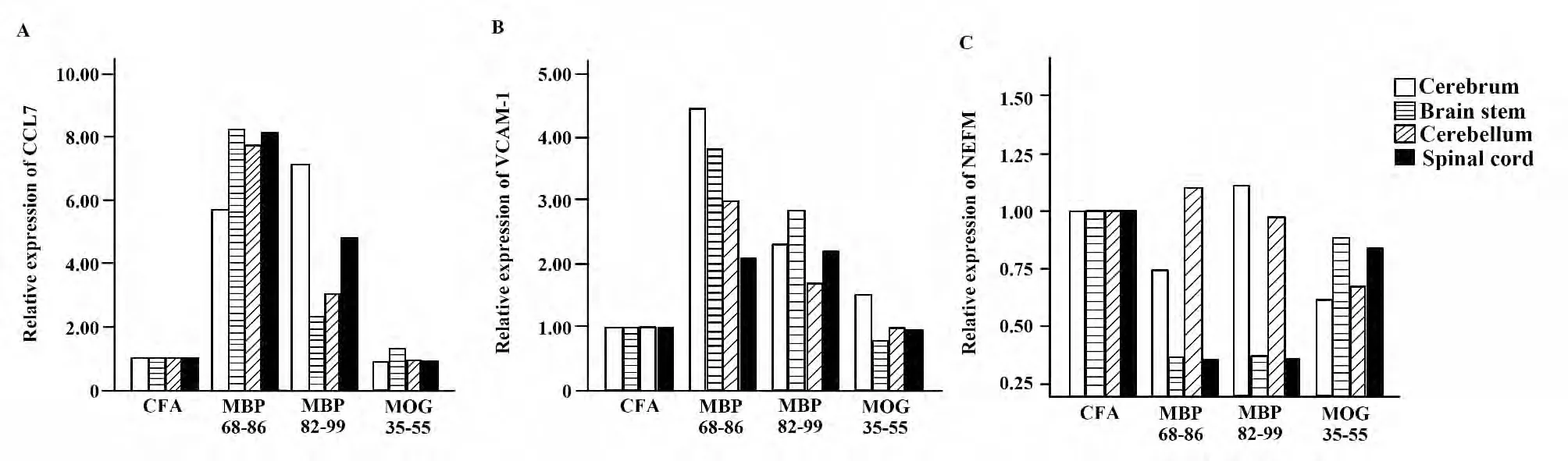
图4 各组大鼠不同部位神经组织的CCL-7、VCAM-1、NEFM 基因表达调节Fig.4 Relative mRNA expression of CCL-7,VCAM-1 and NEFM
2.3 各组大鼠不同部位神经组织CCL-7、VCAM-1、NEFM 基因的表达调节情况 CCL-7 mRNA 表达情况(图4A):MBP 组大鼠的大脑、脑干、小脑和脊髓该基因表达上调,MOG 组大鼠各部位的基因表达无上下调节。
VCAM-1 mRNA 表达情况(图4B):MBP68-86组大鼠的大脑、脑干、小脑、脊髓和MBP82-99 组大鼠的大脑、脑干和脊髓该基因表达上调。NEFM mRNA 表达情况(图4C):MBP68-86 组和MBP82-99组大鼠的脑干和脊髓该基因表达上下调。
2.4 炎性细胞浸润数目与CCL-7、VCAM-1、NEFM基因的相对表达之间的关系 MBP82-99 和MBP 68-86 两组大鼠脊髓的炎性细胞浸润数目/cm2与CCL-7、VCAM-1 的mRNA 表达水平呈正相关(R1=0.52,R2=0.17),脊髓和脑干NEFM 的mRNA 水平与炎性细胞浸润数目/cm2呈负相关(r=-0.61)。
3 讨论
MS 的临床表现和病理学特征具有高度异质性[1]。MS 的临床表现和预后均与神经系统的炎性细胞浸润部位有关,但炎性细胞选择性浸润不同的神经组织部位的机制尚不明确。我们推测可能与不同的磷脂肽段受免疫攻击有关。因此在本研究中,我们采用不同的磷脂肽段免疫Lewis 大鼠,并观察其病理学表现,发现不同的磷脂肽段可以诱发不同的病理学特征。
本研究发现3 种肽段免疫Lewis 大鼠后,病灶分布不同:MBP82-99、MBP68-86 两组大鼠的脊髓、大脑、脑干和小脑中可发现炎性细胞浸润灶,但是仅在MOG35-55 组大鼠的脊髓中发现炎性病灶,且炎性细胞浸润程度不一致,但是不同磷脂肽段诱导不同的炎性病灶分布的原因尚不明确。本研究使用具有相同遗传背景的大鼠,仍发现病灶分布的差异,因此可除外遗传背景的影响。免疫所用的肽段具有不同的溶解性和生物利用度可能是另外一个影响肽段致脑炎活性的原因。Lassmann 等研究发现将不同磷脂肽段特异性T 细胞转移至Lewis 大鼠体内,诱导的不同部位神经组织的病灶,是由于所用磷脂肽段不同所致[7]。因此,T 细胞的抗原特异性可能可以部分解释EAE 的不同临床表现和病理表现。
本研究使用200 μg MOG35-55 免疫Lewis 大鼠,观察8 周,并未发现模型大鼠表现出相应的临床体征,且仅在脊膜下观察到炎性细胞浸润,与既往研究不一致。既往曾有研究使用200 μg MOG35-55 免疫Lewis 大鼠,在免疫后2 周左右表现出体重下降和后肢无力。虽然Adelmann[5]观察到MOG 诱导的EAE 神经系统的炎症,但是同本研究一样,均未发现相应的临床体征。Ichikawa[15]发现MOG35-55 诱导的神经组织炎性细胞浸润主要集中在侧脑室周围白质、丘脑、脑干、小脑和脊髓。然而,在我们的研究中,仅在脊膜下发现炎性细胞浸润,脊髓实质和其余部位神经组织均未发现炎性细胞。随后的RT-PCR实验发现神经组织中趋化因子和黏附分子的水平基本无上调,可能与之相关。本实验动物临床体征、病理学特征与既往研究不同的原因可能在于:与其他研究所使用的Lewis 大鼠的克隆不同。
本研究通过免疫组织化学方法并未发现各组大鼠神经组织存在轴索损害,但是在神经组织中炎性浸润最明显的部位——脊髓和脑干的NEFM mRNA表达水平下降,且相关分析显示其mRNA 水平与炎性浸润灶的数目呈负相关,由于神经丝是轴索的组成成分之一,实验结果提示神经组织炎症反应明显时可能引起神经丝合成障碍,从而导致轴索受累。
CCL-7 和VCAM-1 在EAE 的诱导过程中非常重要。本研究发现MBP82-99 和MBP68-86 免疫大鼠的神经组织中CCL-7 和VCAM-1 的mRNA 水平增加,而且炎性病灶数目与两者的mRNA 水平呈正相关,进一步证明了趋化因子和黏附分子在外周炎性细胞通过血脑屏障进入CNS 过程中的重要性。
该研究发现不同磷脂蛋白诱导Lewis 大鼠神经组织炎性细胞浸润的程度和分布不同,且由于每个MS 患者受累的神经组织不尽相同,部分以脊髓和延髓受累为主,部分患者的侧脑室周围白质和脑干受累,因此给予我们的提示是:对不同自身磷脂肽段的攻击可能导致了不同神经部位神经组织的受累。
[1]Sato F,Martinez NE,Omura S,et al.Heterogeneity versus homogeneity of multiple sclerosis[J].Expert Rev Clin Immunol,2011,7(2):165-167.
[2]Alastair Compston,Alasdair Coles.Multiple slcerosis[J].Lancet,2008,372:1502-1517.
[3]王维治.神经系统脱髓鞘疾病[M].北京:人民卫生出版社,2011:162-173.
[4]Lucchinetti C F,Bruck W,Rodriguez M,et al.Distinct patterns of multiple sclerosis pathology indicates heterogeneity on pathogenesis[J].Brain Pathol,1996:6(3):259-274.
[5]Adelmann M,Wood J,Benzel I,et al.The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG)induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat[J].J Neuroimmunol,1995,63(1):17-27.
[6]Berger T,Weerth S,Kojima K,et al.T-cells against MBP,MOG,MAG,GFAP and S100β induce antigen specific pathological features of nervous system inflammation in EAE[J].J Neuroimmunol,1995,56-63,Supplement 1(0):29.
[7]Berger T,Weerth S,Kojima K,et al.Experimental autoimmune encephalomyelitis:the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system[J].Lab Invest,1997,76(3):355-364.
[8]Abromson-Leeman S,Bronson R,Luo Y,et al.T-cell properties determine disease site,clinical presentation,and cellular pathology of experimental autoimmune encephalomyelitis[J].Am J Pathol,2004,165(5):1519-1533.
[9]Mao YS,Lu CZ,Wang X,et al.Induction of experimental autoimmune encephalomyelitis in Lewis rats by a viral peptide with limited homology to myelin basic protein[J].Exp Neurol,2007,206(2):231-239.
[10]张海琴,李坤成,于春水,等.3.0T 临床型MR 多发性硬化大鼠颅脑成像的初步研究[J].医学影像学杂志,2010,20(2):251-253.
[11]Archambault AS,Sim J,Mccandless EE,et al.Region-specific regulation of inflammation and pathogenesis in experimental autoimmune encephalomyelitis[J].J Neuroimmunol,2006,181(1-2):122-132.
[12]Kuerten S,Kostova-Bales DA,Frenzel LP,et al.MP4-and MOG35-55-induced EAE in C57BL/6 mice differentially targets brain,spinal cord and cerebellum[J].J Neuroimmunol,2007,189(1-2):31-40.
[13]王 琼,高波廷,王 伟,等.小鼠实验性自身免疫性脑脊髓炎的病理变化[J].卒中与神经疾病,2006,13(2):97-99,102.
[14]Jovanova-Nesic K,Shoenfeld Y.MMP-2,VCAM-1 and NCAM-1 expression in the brain of rats with experimental autoimmune encephalomyelitis as a trigger mechanism for synaptic plasticity and pathology[J].J Neuroimmunol,2006,181 (1-2):112-121.
[15]Ichikawa M,Johns T G,Liu J,et al.Analysis of the fine B cell specificity during the chronic/relapsing course of a multiple sclerosis-like disease in Lewis rats injected with the encephalitogenic myelin oligodendrocyte glycoprotein peptide 35-55[J].J Immunol,1996,157(2):919-926.
