抗体与小分子药物一体化的完美“联姻”
——解析抗体-药物偶联物之定点偶联
2015-02-10李晨晨王旻张娟
李晨晨,王旻,张娟
(中国药科大学生命科学与技术学院,江苏 南京 210009)
抗体与小分子药物一体化的完美“联姻”
——解析抗体-药物偶联物之定点偶联
李晨晨,王旻,张娟*
(中国药科大学生命科学与技术学院,江苏 南京 210009)
基于单抗的靶向疗法已成为各种癌症的重要治疗手段,抗体与小分子药物一体化的联姻——抗体—药物偶联物(antibody-drug conjugates,ADC)新药获得了突破性进展。传统ADC是将药物与抗体的赖氨酸残基或链间二硫键还原而产生的半胱氨酸残基相偶联而形成,其稳定性差,易发生聚集,且其中药物易脱落而产生非治疗性毒副作用。而应用近年发展起来的定点偶联技术所获ADC,除均一性好外,还保留了母体单抗的药动学性质,毒副作用也远低于具有相同偶联比的传统ADC,极有可能发展成为新一代重磅药物。综述4种ADC定点偶联方法。
抗体—药物偶联物;定点偶联;反应性半胱氨酸;非天然氨基酸;酶法;二硫键改造
抗体药物以高特异性、高稳定性和低毒性等优点成为现代生物药物发展的中流砥柱,几乎每一株抗体都被寄予希望成为“重磅药物”(blockbuster drug)而诞生。在抗肿瘤治疗中,抗体药物也已成为标准疗法,但其单独使用的疗效常常不尽如人意[1-3]。近年来,抗体与小分子药物的一体化联姻——抗体-药物偶联物(antibodydrug conjugates,ADC)新药获得了突破性进展,极有可能成长为抗体肿瘤疗法中新一代重磅药物。
ADC药物是将细胞毒性药物通过小分子连接臂(linker)连接至单抗,依靠单抗的独特定位作用,使药物结合并杀死癌细胞,弥补了细胞毒性药物副作用偏大和单抗疗效偏弱等缺陷[1-5]。表1展示了已上市及在研的部分ADC药物信息。
传统的ADC是将药物偶联在抗体的赖氨酸(Lys)残基或是链间二硫键还原而产生的半胱氨酸(Cys)残基上。由于一个抗体大约含有40个Lys,通过Lys偶联而形成的ADC,其药物-抗体的偶联比(drug-to-antibody ratios, DAR)为0~8,但通过打开的链间二硫键偶联,则会破坏抗体的完整性[1-5]。因此,由经典方法产生的ADC是一个混合物,且不稳定。近年来,有研究者运用基因工程技术将抗体进行改造,从而实现抗体与药物的定点偶联,由此所得ADC不仅具有十分均一的DAR,而且还表现出极好的体内药动学性质,大大提高了治疗效果[6]。本文综述抗体与药物的4种定点偶联方法:引入反应性Cys或非天然氨基酸、酶法及对二硫键进行改
造,并对这些定点偶联技术进行分析,期望对ADC药物 的研究和开发提供理论指导。
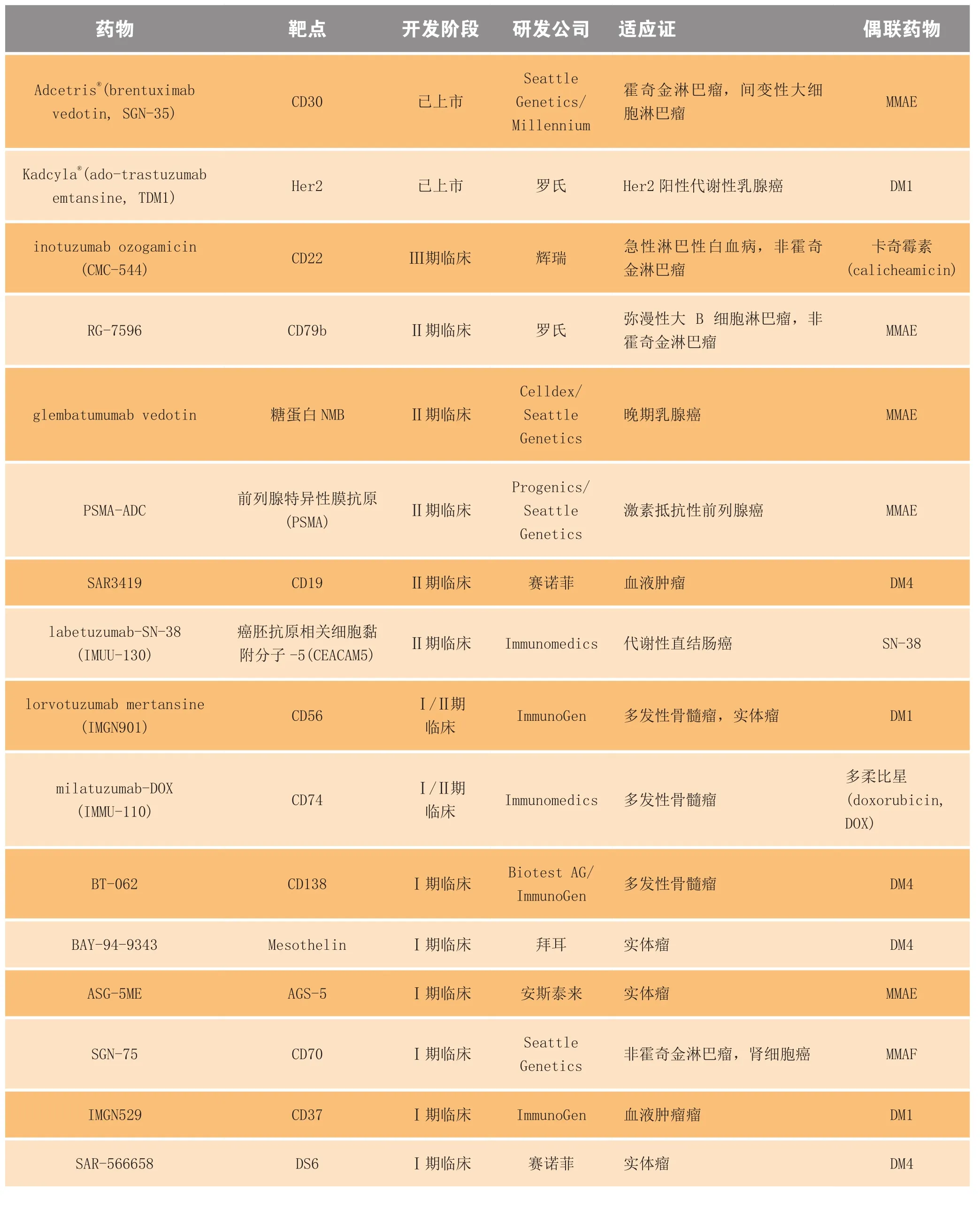
表1 已上市及处于临床研究阶段的部分ADC药物Table1 Some ADCs launched or in clininal trials
1 引入反应性半胱氨酸实现定点偶联
研究发现,将抗体分子中某一氨基酸残基突变成Cys,再利用其与药物进行特异性偶联而合成ADC,这样便可消除链间二硫键破坏所造成的影响。然而,如果突变位点的设计不恰当,将会形成错误的链内或链间二硫键[7-8]。Junutula等[7]研究开发了PHESELECTOR (Phage ELISA for Selection of Reactive Thiols) 技术(见图1),即首先选出一些完全或部分暴露于抗体表面的氨基酸[如缬氨酸(Val)、丙氨酸(Ala)、丝氨酸(Ser)等] 残基,将其突变为Cys;再利用噬菌体展示技术,将该Fab片段突变体展现于噬菌体表面,与马来酰亚胺-PEO-生物素反应;然后,经ELISA方法筛选得到既保持抗原结合能力又能通过引入的Cys残基与生物素(Biotin)成功偶联(能结合链霉亲和素)的突变体。同时,该研究团队以曲妥珠单抗(trastuzumab)的Fab片段为基础,筛选出位于重链(HC)或轻链(LC)上符合上述条件的3个突变位点:HC-A114、HC-A175和LC-V110,并应用此技术获得含有反应性Cys的IgG突变体THIOMAB,其与药物的偶联产物称为TDC(THIOMAB-drug conjugates)。
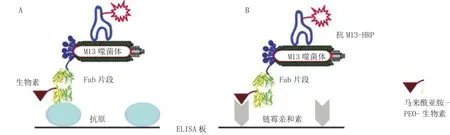
图1 PHESELECTOR 技术示意图Figure 1 Schematic representation of PHESELECTOR
随后,Junutula研究团队利用THIOMAB平台,将曲妥珠单抗中HC-A114位点突变为Cys,并通过含有BMPEO的连接臂偶联DM1,成功构建了TDC,即thiotrastuzumab-mepo-DM1(thio-TDM1)。LC/MS分析显示,thio-TDM1的平均DAR为1.8,其中DAR为2的偶联物占比达90%。且体内外实验显示,尽管thio-TDM1中偶联的DM1数量较已上市的Kadcyla®(TDM1)要少(330vs560 μg·mm-2),对于过表达Her2的SK-BR-3细胞,这两种偶联物表现出相似的细胞表面抗原结合能力和细胞毒性;在Fo5小鼠乳腺癌模型(乳腺癌上皮细胞过表达人Her2)中,两种偶联物在相同的给药剂量(10 mg·kg-1)下,可产生相似的疗效;用于大鼠和猕猴时,在相同的给药剂量(47 mg·kg-1)下,thio-TDM1产生的毒性远小于TDM1[9]。此外,将抗MUC16抗体中HC-A114位点突变为Cys后,与MMAE偶联,所产生的TDC也有类似优点[10]。而在抗CD33抗体中引入反应性Cys后,与DNA交联剂吡咯苯二氮 二聚体偶联,形成的TDC(SGN-CD33A)平均DAR为1.9,其中DAR为2的偶联物占比达95%,该TDC药物现已进入Ⅰ期临床试验[11-12]。
Shen等[13]在曲妥珠单抗上选取了3个突变位点:HC-A114、LC-V205和Fc-S396,用于考察不同偶联位点对TDC稳定性的影响。这3个位点的区别在于溶剂可及性和周围电荷环境的不同:Fc-S396位于最易触及的部位,而HC-A114和LC-V205位点则呈部分暴露;LCV205位点周围呈正电荷环境,而HC-A114和Fc-S396则处于相对中性的环境。当将MMAE分别与含有上述不同突变位点的THIOMAB偶联时,所得3种TDC具有相似的DAR(1.7~1.9)。体外活性实验表明,这3种TDC的抗原结合能力、SK-BR-3细胞(过表达Her2)内化程度和细胞毒性均无差异,但是它们的血浆稳定性、体内半衰期以及体内活性却相差极大,其中,偶联于LCV205位点的TDC稳定性最好,血浆中37 ℃孵育96 h后未见降解。
与常规ADC相比,通过PHESELECTOR技术获得的相应TDC虽然表现出更好的治疗效应,但也存在缺陷:展示在噬菌体表面的Fab片段无需经任何处理即可与马来酰亚胺反应,而全长抗体在哺乳动物细胞中表达时,引入的Cys可以和培养基中谷胱甘肽等含巯基的物质形成二硫键,所以还需将其还原成游离的形式才能进行偶联,但与此同时,抗体的链间二硫键也会被打开,这又需再次氧化以恢复成完整的抗体;而且,在THIOMAB形成的整个过程中,这个额外引入的巯基也有可能在抗体的2个Fab间错误地形成二硫键[9-12]。
2 引入非天然氨基酸实现定点偶联
有研究者利用遗传密码扩充技术,合成一个可以识别终止密码子的tRNA,并设计能催化非天然氨基酸连接在该tRNA上的氨酰tRNA合成酶,构成氨酰tRNA合成酶/tRNA正交对;然后,将抗体的DNA序列中某一氨基酸密码子突变为终止密码子,再协同这一正交对,在细胞内或细胞外合成含有非天然氨基酸的抗体[14]。由于引入的非天然氨基酸含有一些特殊官能团,如叠氮基、酮基、炔基等,致使药物与抗体能更轻易地实现定点偶联。这项技术的关键在于,获得严格正交的分子对,利用其高反应特异性将非天然氨基酸定点引入抗体分子中。
2.1 细胞内翻译引入非天然氨基酸
Axup等[15]以曲妥珠单抗的Fab片段为基础,设计了3个突变体,每个突变体分别在HC或LC的不同位点(HC-A121、LC-S202和LC-K169)引入一个琥珀突变密码子(UAG),并与对乙酰苯丙氨酸(pAcF,1)氨酰tRNA合成酶/tRNA正交分子对在大肠杆菌中共表达,产生相应位置含有pAcF的Fab片段突变体;同时,将正交分子对的DNA序列整合在CHO-K1细胞的基因组中,表达出含有琥珀突变位点(HC-A121)的IgG全长抗体。ESI-MS分析显示,表达出的3个Fab片段和1个全长IgG在突变位点处插入了pAcF,且抗体分子产量均与未引入突变的母体单抗相当。全长抗体突变体上的酮基通过与含有羟胺基团的连接臂形成肟键,从而将MMAF偶联至pAcF位点,得到偶联率达95%、DAR为2的ADC。实验显示,在荷有Her2过表达的MADMB-435细胞(乳腺下种植105个细胞)的C.B-17/SCID小鼠模型中,单剂量给予该ADC(5 mg·kg-1),即可在2周内清除肿瘤,且该ADC在小鼠体内的清除率与母体单抗相当[(0.31±0.03)vs(0.3±0.11)mL·h-1·kg-1]。
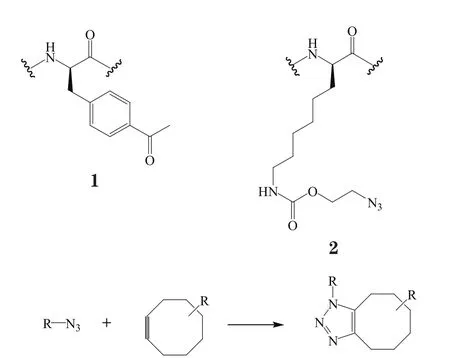
图2 SPAAC 反应Figure 2 SPAAC reaction
变形促进的叠氮-辛炔环加成反应(strain-promoted azide-alkyne cycloaddition,SPAAC)是环八炔基和叠氮基团间的[3+2]环加成反应,可形成三氮唑并环八烷基偶联物(见图2)[16]。该反应无需铜(Ⅰ)的催化,具有高效、条件温和等特点,被广泛应用于多种生物偶联反应。Xiao等[17]在曲妥珠单抗的HC-A121和LC-110位点分别引入琥珀突变密码子(UAG)和赭石突变密码子(UAA),并与2个正交分子对(pAcF氨酰tRNA合成酶/tRNA和N-t-2-叠氮乙氧羰基-L-赖氨酸(AzK,2)氨酰tRNA合成酶/tRNA共同转入Freestyle293-F细胞中,生成在相应位点处分别含有2个非天然氨基酸的曲妥珠单抗突变体——trastuzumabpAcF/AzK。其中引入的非天然氨基酸pAcF和AzK分别含有乙酰基团和叠氮基团,它们可分别与linker-MMAF(含有羟胺基团)和DIBO-AF488(含有炔基)通过肟键和SPAAC反应相连接,形成抗体-药物/荧光素双偶联物(trastuzumab-MMAF/DIBO)。LC/MS分析表明,这2个修饰物的成功偶联率达90%。且在过表达Her2的SK-BR-3细胞表面抗原结合以及内化实验中,双偶联产物trastuzumab-MMAF/DIBO完全保持了母体单抗的活性。
上述偶联方法虽可在抗体的特定部位引入能参与偶联反应的特殊官能团,但实现起来同样面临缺陷:由于氨酰tRNA合成酶/tRNA对往往不是严格正交,故可能会在突变位点引入结构相似的其他天然氨基酸,或使翻译提前终止;而且,含有非天然氨基酸的氨酰tRNA也能干扰内源性终止密码子,造成细胞自身蛋白的错误翻译;此外,这些非天然氨基酸本身多数具有细胞毒性。为此,一些研究者开始探索利用无细胞蛋白质表达系统来引入非天然氨基酸。
2.2 细胞外翻译引入非天然氨基酸
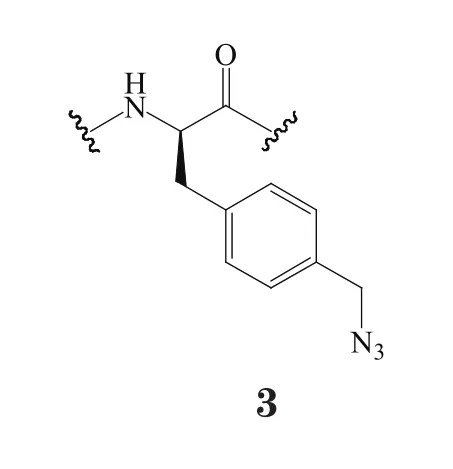
无细胞蛋白质表达系统是指利用细胞抽提物中的蛋白质合成元件、并添加底物和能量所构成的以mRNA为模板的体外翻译系统。对叠氮基甲基-L-苯丙氨酸(pAMF,3)是在苯丙氨酸(Phe)基础上引入一个叠氮基团而形成的非天然氨基酸,其可高效地进行SPAAC反应,但多项研究显示,不能有效构建pAMF氨酰tRNA合成酶/tRNApAMF正交对。Zimmerman等[18]先是利用体外翻译系统作为一个高通量筛选平台,从1 760个克隆中筛选出一株高保真性的pAMF氨酰tRNA合成酶B03;随后,在曲妥珠单抗的HC-S136位点引入琥珀突变密码子(UAG),并利用筛选出的B03酶、tRNA以及具适合比例的氧化型谷胱甘肽(GSSG)/还原型谷胱甘肽(GSH),在大肠杆菌抽提物中对该抗体突变体进行无细胞蛋白质表达。结果,由此得到的抗体中,可见pAMF特异性的插入突变位点,而未见有其他类似结构的非天然氨基酸[如Phe、酪氨酸(Tyr)等]插入,同时也未出现翻译提前终止的截短体。而且,该抗体突变体中的pAMF基团能与含有DBCO的MMAF-linker通过SPACC反应连接,偶联形成的ADC纯度大于90%。
目前,利用大肠杆菌抽提物进行体外翻译的研究已较为成熟。研究表明,原核细胞的表达系统不能对抗体进行糖基化修饰,而且表达的产物中带有内毒素;相比之下,真核来源的体外翻译系统则更适合进行抗体大分子的表达,且真核细胞裂解物中含有源于内质网的微粒小囊泡,其中富含能辅助蛋白质正确折叠、促进二硫键形成的多种分子伴侣以及参与糖基化修饰的酶类[18-20]。Stech等[20]尝试利用昆虫细胞裂解物合成了含有非天然氨基酸的scFv抗体片段,并成功将其用于与药物的偶联,但偶联产物中仍保留有N端信号肽序列。由此看来,真核体外翻译系统虽有利于抗体的非天然氨基酸修饰,但此体外翻译平台尚有待进一步的开发和完善。
3 通过酶法实现定点偶联
此方法是指,通过基因工程技术,人为地致使抗体中产生能被某些酶所识别的相关氨基酸序列,再利用酶对底物的特异性,将其中的特定氨基酸残基进行改造修饰,从而实现定点偶联[21]。
3.1 利用甲酰甘氨酸生成酶
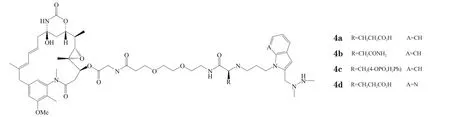
甲酰甘氨酸生成酶(formylglycine-generating enzyme,FGE)可以识别一个五肽序列CXPXR[C、P、R分别为Cys、脯氨酸(Pro)和精氨酸(Arg),X为Ser、Ala、苏氨酸(Thr)或甘氨酸(Gly)],并将其中的Cys氧化成甲酰甘氨酸(fGly)[22],而产生的甲酰基团可以通过HIPS反应与药物形成稳定的C- C键,将药物偶联在抗体上(见图3)。 Drake等[23]通过该途径,在曲妥珠单抗的C端引入LCTPSR序列,并于整合有FGE基因的CHO细胞中表达,得到的抗体在相应位点含有fGly基团。随后,该研究团队通过引入不同的氨基酸[谷氨酸(Glu)、天冬酰胺(Asn)和磷酸Tyr]对连接臂进行修饰,并与DM1偶联,产生不同ADC(4a、4b、4c、4d),考察不同连接臂对偶联产物的影响[24]。结果显示,ADC产物的偶联率达90%,平均DAR为1.6,单体率为95%,其中,ADC 4d的血浆稳定性最好,在人血浆(37 ℃)中1周和2周的降解率分别为3%和17%;通过这些极为相似的连接臂偶联所得ADC其体内外活性却存在很大差异,在NCI-N87荷瘤鼠模型中, ADC 4b对大体积瘤体(瘤体积大于400 mm3)的抑制活性显著高于ADC 4a,且其起效也快。

图3 通过FGE和HIPS反应进行的药物与抗体上氨基酸共价连接Figure 3 Covalent binding of drug and the amino acid in antibody through FGE and HIPS reaction
3.2 利用谷氨酰胺转移酶
谷氨酰胺转移酶(transglutaminase,TG)可以识别LLQGA五肽序列[L、Q、G、A分别为亮氨酸(Leu)、谷氨酰胺(Gln)、Gly和Ala],并能催化其中Gln的γ-酰胺基团与含有伯胺的化合物形成异肽链(见图4)。
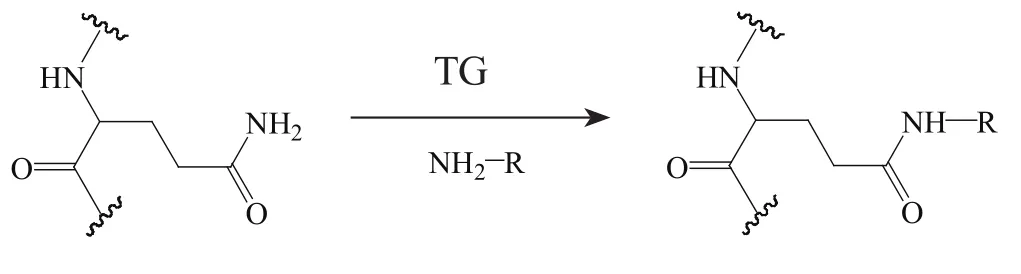
图4 TG催化的伯胺与Gln上γ-酰胺基团共价连接Figure 4 Covalent binding of primary amine and the γ-carbonylamid group in Gln by TG catalysis
Farias等[25]将抗M1S1抗体中HC和LC的C端分别插入LLQGA和GGLLQGA(G、L、Q、A分别为Leu、Gln、Gly和Ala)序列,并基于TG的催化作用与MMAD偶联,得到的ADC其平均DAR分别为1.9和1.8,单体率分别为99%和98.5%。结合LC-MS/MS分析产生的高分辨率肽段图谱和系统性片段化分析技术的考察,发现上述所得ADC中约有1.3%的抗体在HC-Q295残基处形成了额外的偶联位点,原因可能是TG对底物的要求并不严格而将去糖基化的抗体上HC-Q295位点视作其底物所致,而正常抗体由于HC-N297位点会发生糖基化修饰而阻碍TG对附近HC-Q295位点的识别。于是,该研究小组将抗体上HC-Q295位点突变为HC-N295,并再次对其偶联产物进行分析,结果表明这一脱靶偶联效应消失。
对于TG介导的催化反应,由于疏水性的药物是溶解在有机溶剂中的,且其空间位阻较大,这些都不利于TG的催化;相比之下,水溶性小分子衔接物则能实现高效连接。鉴于此,Dennler等[26]利用N-糖酰胺酶F(PNGaseF)将曲妥珠单抗去糖基化后暴露出HC-Q295位点,然后利用含有2个官能团的小分子衔接物(一端为伯胺基团,另一端为叠氮基团),在TG的催化下,先在抗体的此位点引入化学反应性官能团,再将含环八炔基药物-linker(DOBC-PEG4-vc-PAB-MMAE)通过SPAAC反应进行偶联(见图5),由此经酶-化学法两步形成的ADC其偶联效率得以提高,仅用1.25倍剂量的药物,即可达到100%(DRA为2.0)的偶联率,且均一性极好。
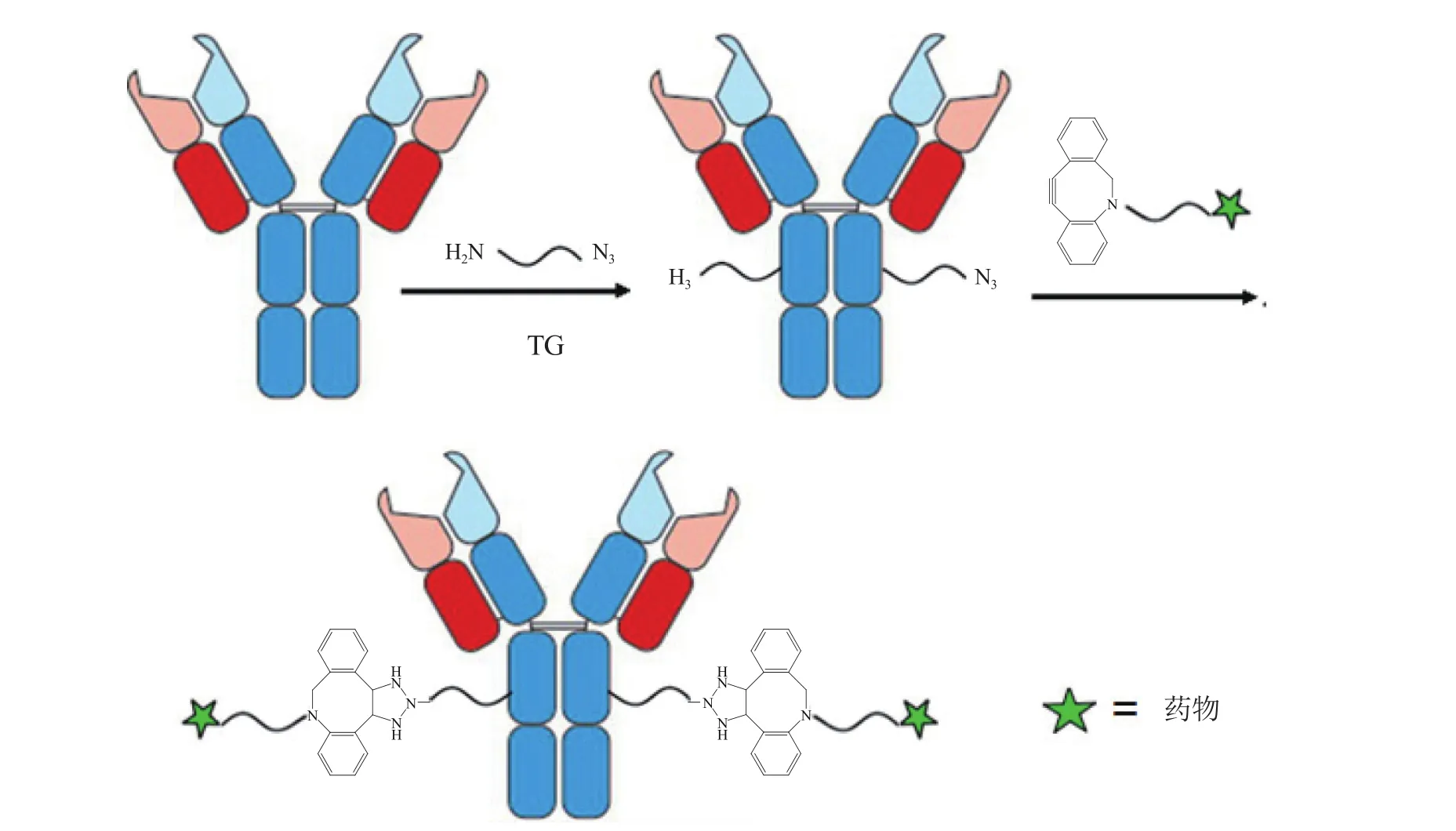
图5 酶-化学法两步定点偶联形成ADCFigure 5 Formation of ADC through two-step site-specific conjugation by chemo-enzymatic approach
最近,Lhospice等[27]将抗CD30抗体cAC10(Adcetris®的母体单抗)上N297位点突变为Q297,这样的抗体自然不会发生糖基化,但却会在HC上形成2个TG催化位点(Q295和Q297),因此,通过该法改造后的一个抗体能偶联4个药物分子。动物体内放射性同位素实验显示,与Adcetris®相比,由改造后抗体形成的ADC在肿瘤部位有较高的摄取率[即注射后24 h每克肿瘤组织的放射性同位素含量:(17.84±2.2)%vs(10.5±1.8)%],而对正常组织的毒性更低。且动物模型实验表明,其在单剂量为0.3 mg·kg-1时的药效略高于Adcetris®。
经TG催化处理后,去糖基化抗体上HC-Q295残基即可作为ADC偶联位点。然而,由于缺少糖基化修饰的抗体不能执行Fc段功能,且不能与FcRn受体结合,因此其抗体功能会削弱且体内循环半衰期也会缩短[28]。但对于ADC来说,由于糖基侧链可与内皮细胞以及肾脏细胞表面的受体结合,故携带至相应部位的毒性药物会造成非特异性损伤。为此,抗体部分的糖基化修饰对形成的ADC的影响仍有待于进一步研究。
4 通过二硫键改造实现定点偶联
当抗体的链间二硫键被打开后,还原的Cys可以通过和双反应性试剂反应,将两条链重新连接起来,即以这个双反应性试剂替换传统的链间二硫键。据此,利用连接有药物的双反应性试剂,可将药物定位于抗体的二硫键位点,形成ADC(见图6)。而且,双反应性试剂的分子质量小,不足以影响抗体分子的空间结构。下面分别介绍2种双反应性试剂在对抗体链间二硫键进行改造而实现定点偶联并形成ADC中的应用。
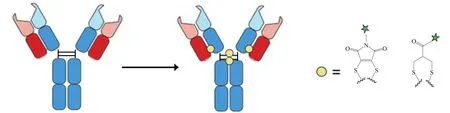
图6 通过二硫键改造定点偶联形成ADCFigure 6 Formation of ADC through site-specific conjugation by disulfide re-bridge approach
4.1 利用新一代马来酰亚胺试剂
通过亲电基团取代马来酰亚胺的3、4位,形成新一代马来酰亚胺试剂(next generation maleimide,NGM),其可构成药物-linker(5),并作为双反应性试剂,能与亲核性的Cys反应,从而将2条具有Cys的抗体单链桥连起来,并致使所携药物与抗体偶联,形成ADC[29-30]。Schumacher等[31]利用此方法,将DOX与曲妥珠单抗偶联,结果,CE-SDS分析显示,所得ADC的产率为52% ~ 69%,完整抗体的比例占74%;实验还发现,以硒酚为还原剂时,可特异性地将DOX偶联在Fab片段的二硫键上,且不改变两条HC间的二硫键,从而得到DAR为2的ADC,这样的偶联方式可以将疏水药物在空间上分开,增加ADC的稳定性。
4.2 利用双烷基化试剂
利用双烷基化试剂构建的药物-linker(6)中对甲基苯磺酰基是很好的离去基团,可发挥双反应性试剂作用,其在反应过程中,能生成含有双键的中间体,并与2个Cys发生加成反应,形成三碳桥而桥连抗体的2条单链,最终偶联得到ADC(见图7)。Badescu等[32]利用该方法,将MMAE通过一个含24个PEG单位的分子链分别偶联在曲妥珠单抗的Fab片段和全长抗体上,结果,实验显示,MMAE与Fab片段的偶联产物,纯度高(95%以上),且无沉淀产生;MMAE与全长抗体偶联得到的ADC平均DAR为2.3,其中DAR为4的偶联产物占比达78%。与NGM桥联法相比,该法的最大优势在于能获得稳定性很好的ADC,其所得ADC于37 ℃血清中孵育5 d后未见降解,而在相同条件下,NGM偶联相同药物所获ADC的降解率却可达50%。
利用双反应性试剂进行定点偶联,不需在DNA水平上对抗体进行改造,且偶联产物的DAR均一性很好。更重要的是,经ELISA检测表明,上述2种双反应性试剂偶联方法所获ADC均完全保留了与抗原的结合能力,提示,改造后的二硫键未对抗体的空间结构造成影响。
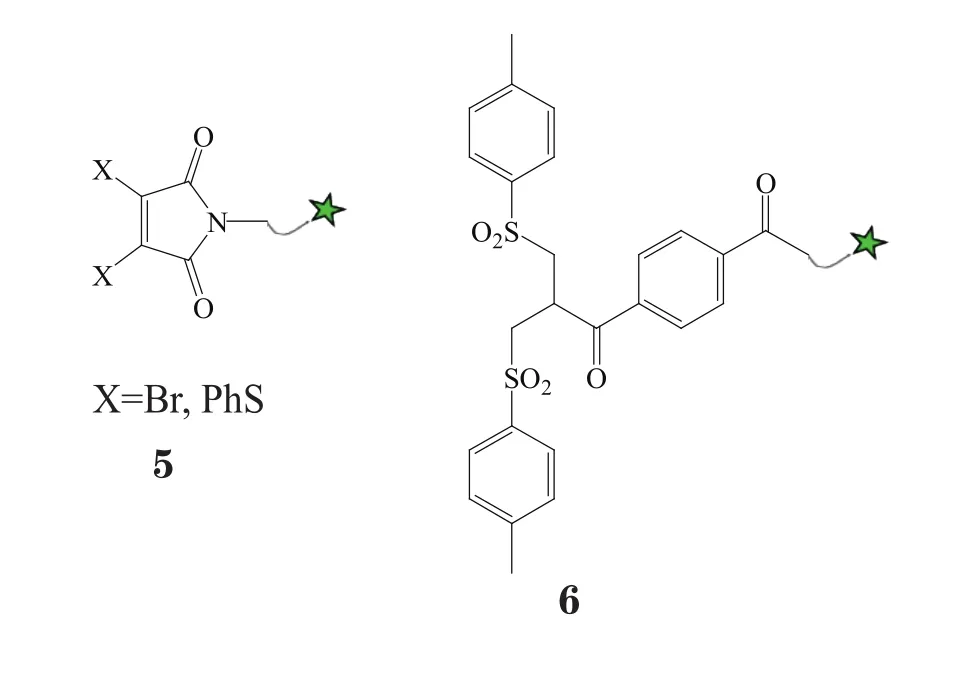
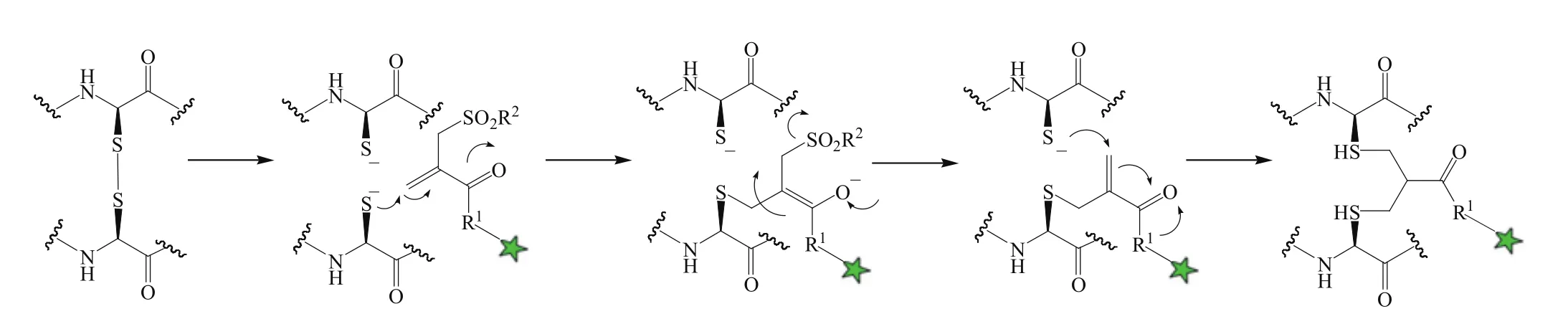
图7 利用双烷基化试剂将药物定位于二硫键处Figure 7 Conjugation of a drug to a disulfide bridge using bis-alkylating reagent
5 结语
传统方法制得的ADC,其批次间差异较大,很难制定一个对其进行药物分析以及体内外活性考察的评价标准;此外,小分子药物具有强疏水性,偶联较多数量药物(DAR为4以上)的ADC极易发生聚沉,稳定性降低。体内实验数据显示,相比于传统ADC,定点偶联产生的ADC其药动学性质极好,大大降低了由于药物脱落而导致的非治疗性毒副作用,因而其有更宽的治疗窗。表2总结了本篇综述所介绍的4种ADC定点偶联方法的各自特点,它们虽然各自都有不同的优缺点,但较之传统的非定点偶联技术均有其不可比拟的优势,定点偶联将成为ADC未来发展的必然趋势。
今后对ADC定点偶联技术的研究,还可进一步探讨连接臂、偶联位点以及偶联药物对ADC的影响。
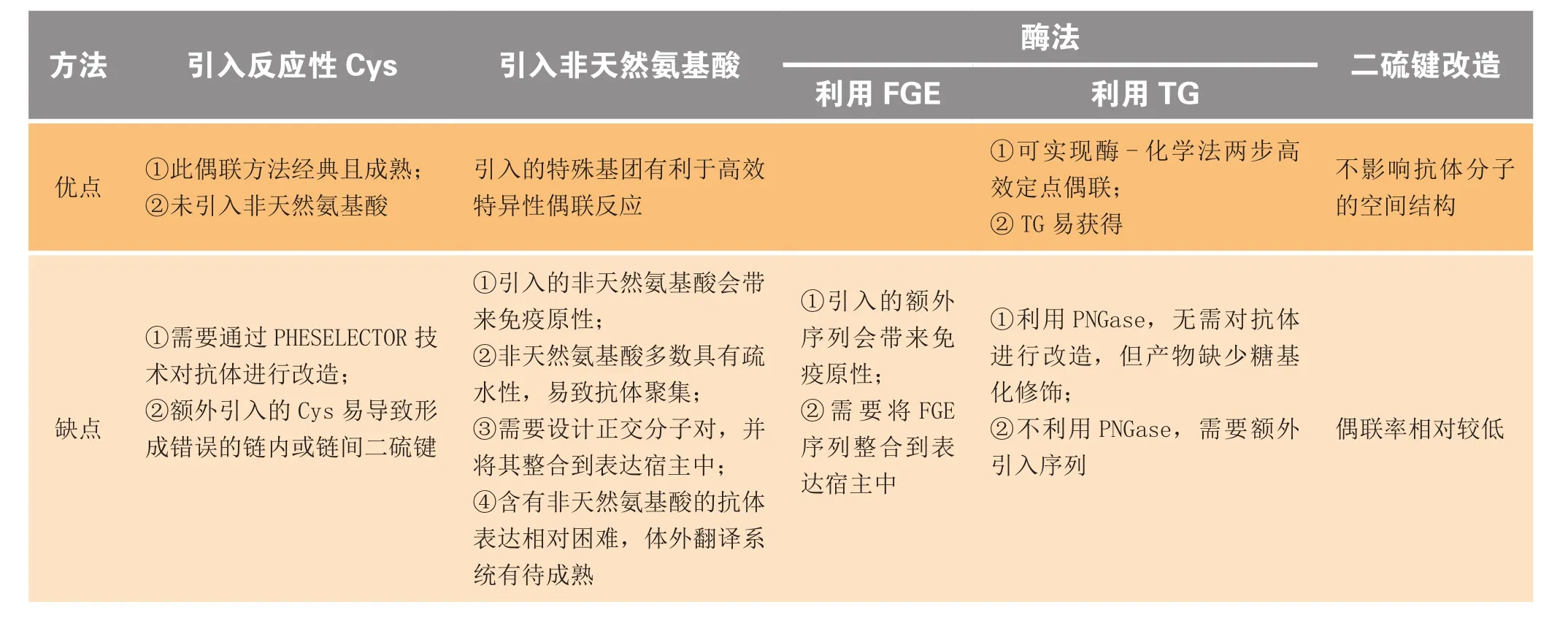
表2 4种ADC定点偶联方法的特点Table2 Characteristics of 4 site-specific conjugation methods for ADC
[1]Behrens C R, Liu B.Methods for site-specific drug conjugation to antibodies[J].MAbs, 2014, 6(1): 46-53.
[2]Tian F, Lu Y, Manibusan A,et al.A general approach to site-specific antibody drug conjugates[J].Proc Natl Acad Sci USA, 2014, 111(5): 1766-1771.
[3]Panowksi S, Bhakta S, Raab H,et al.Site-specific antibody drug conjugates for cancer therapy[J].MAbs, 2014, 6(1):34-45.
[4]Agarwal P, Bertozzi C R.Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development[J].Bioconjug Chem, 2015, 26(2):176-192.
[5]van Vught R, Pieters R J, Breukink E.Site-specifc functionalization of proteins and their applications to theraputic antibodies[J].Comput Struct Biotechnol J, 2014, 9: e201402001.
[6]Beck A, Reichert J M.Antibody-drug conjugates: present and future[J].MAbs, 2014, 6(1): 15-17.
[7]Junutula J R, Bhakta S, Raab H,et al.Rapid identifcation of reactive cysteine residues for site-specific labeling of antibody-Fabs[J].J Immunol Methods, 2008, 332(1/2): 41-52.
[8]Shen B Q, Xu K, Liu L,et al.Conjugation site modulates thein vivostability and therapeutic activity of antibody-drug conjugates[J].Nat Biotechnol, 2012, 30(2): 184-189.
[9]Junutula J R, Flagella K M, Graham R A,et al.Engineered thiotrastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer[J].Clin Cancer Res, 2010, 16(19): 4769-4778.
[10]Junutula J R, Raab H, Clark S,et al.Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index[J].Nat Biotechnol, 2008, 26(8): 925-932.
[11]Kung Sutherland M S, Walter R B, Jeffrey S C,et al.SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML[J].Blood, 2013, 122(8): 1455-1463.
[12]Seattlegentics.SGN-CD33A[EB/OL].[2015-02-27].http://www.seattlegenetics.com/SGN-CD33A.
[13]Shen B Q, Xu K, Liu L,et al.Conjugation site modulates thein vivostability and therapeutic activity of antibody-drug conjugates[J].Nat Biotechnol, 2012, 30(2): 184-189.
[14]Javahishvili T, Manibusan A, Srinagesh S,et al.Role of tRNA orthogonality in an expanded genetic code[J].ACS Chem Biol, 2014, 9(4): 874-879.
[15]Axup J Y, Bajjuri K M, Ritland M,et al.Synthesis of site-specific antibody-drug conjugates using unnatural amino acids[J].Proc Natl
Acad Sci USA, 2012, 109(40): 16101-16106.
[16]Lallana E, Riguera R, Fernandez-Megia E.Reliable and efficient procedures for the conjugation of biomolecules through Huisgen azidealkyne cycloadditions[J].Angew Chem Int Ed Engl, 2011, 50(38): 8794-804.
[17]Xiao H, Chatterjee A, Choi S H,et al.Genetic incorporation of multiple unnatural amino acids into proteins in mammalian cells[J].Angew Chem Int Ed Engl, 2013, 52(52): 14080-14083.
[18]Zimmerman E S, Heibeck T H, Gill A.Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system[J].Bioconjug Chem, 2014, 25(2): 351-361.
[19]Quast R B, Claussnitzer I, Merk H,et al.Synthesis and site-directed fluorescence labeling of azido proteins using eukaryotic cell-free orthogonal translation systems[J].Anal Biochem, 2014, 451: 4-9.
[20]Stech M, Hust M, Schulze C,et al.Cell-free eukaryotic systems for the production, engineering, and modification of scFv antibody fragments[J].Eng Life Sci, 2104, 14(4): 387-398.
[21]Cho H, Jaworski J.Enzyme directed formation of un-natural sidechains for covalent surface attachment of proteins[J].Colloids Surf B Biointerfaces, 2014, 122: 846-850.
[22]Appel M J, Bertozzi C R.Formylglycine, a post-translationally generated residue with unique catalytic capabilities and biotechnology applications[J].ACS Chem Biol, 2015, 10(1): 72-84.
[23]Drake P M, Albers A E, Baker J,et al.Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifcally to different antibody regions with distinctin vivoefficacy and PK outcomes[J].Bioconjug Chem, 2014, 25(7): 1331-1341
[24]Albers A E, Garofalo A W, Drake P M,et al.Exploring the effects of linker composition on site-specifically modified antibody-drug conjugates[J].Eur J Med Chem, 2014, 88: 3-9.
[25]Farias S E, Strop P, Delaria K,et al.Mass spectrometric characterization of transglutaminase based site-specific antibody-drug conjugates[J].Bioconjug Chem, 2014, 25(2): 240-250.
[26]Dennler P, Chiotellis A, Fischer E.Transglutaminase-based chemoenzymatic conjugation approach yields homogeneous antibody-drug conjugates[J].Bioconjug Chem, 2014, 25(3): 569-578.
[27]Lhospice F, Brégeon D, Belmant C,et al.Site specific conjugation of monomethyl auristatin E to anti CD30 antibodies improves their pharmacokinetics and therapeutic index in rodent models[J].Mol Pharm, 2015, 12(6): 1863-1871.
[28]Ward E S, Devanaboyina S C, Ober R J.Targeting FcRn for the modulation of antibody dynamics[J].Mol Immunol, 2015 Mar 9.pii: S0161-5890(15)00047-4.doi: 10.1016/j.molimm.2015.02.007.[Epub ahead of print].
[29]Schumacher F F, Nobles M, Ryan C P,et al.In situ maleimide bridging of disulfdes and a new approach to protein PEGylation[J].Bioconjug Chem, 2011, 22(2): 132-136.
[30]Castañeda L, Maruani A, Schumacher F F,et al.Acid-cleavable thiomaleamic acid linker for homogeneous antibody-drug conjugation[J].Chem Commun(Camb), 2013, 49(74): 8187-8189.
[31]Schumacher F F, Nunes J P, Maruani A,et al.Next generation maleimides enable the controlled assembly of antibody-drug conjugates via native disulfde bond bridging[J].Org Biomol Chem, 2014, 12(37): 7261-7269.
[32]Badescu G, Bryant P, Bird M,et al.Bridging disulfdes for stable and defined antibody drug conjugates[J].Bioconjug Chem, 2014, 25(6): 1124-1136.

[专家介绍] 张娟:博士,副教授,硕士生导师。1997年毕业于中国药科大学,2009年获中国药科大学微生物与生化药学博士学位。2007年,赴英国帝国理工学院博士联合培养;2013-2014年,作为公派学者赴美国加州大学洛杉矶分校(UCLA)医学院Morrison教授实验室从事抗骨髓瘤抗体研究工作;中国生化药物杂志编委;入选江苏省“青蓝工程”中青年学术带头人、江苏省第四期“333工程”第三层次培养对象、中国药学会青年生物药物科学家。
张娟副教授主要从事新型抗肿瘤抗体研究和开发工作,基于噬菌体展示抗体文库、杂交瘤及抗体人源化技术获得多株单克隆抗体,并进行创新型双靶点抗体、抗体融合蛋白及抗体偶联药物(ADC)的研究和开发,近5年主持及主要负责5项省部级抗体研究项目,授权抗体专利3项,公开抗体专利5项,发表SCI及中文核心论文40余篇。
Perfect Coupling of Antibodies with Small Molecule Drugs: Site-specifc Conjugation for Antibody-Drug Conjugates
LI Chenchen, WANG Min, ZHANG Juan
(School of Life Science & Technology, China Pharmaceutical University, Nanjing 210009, China)
Antibody based targeted therapy has become more and more popular in anti-cancer therapy.Coupling of antibodies with small molecule drugs——antibody-drug conjugate(ADC) which is a new class of therapeutics has got a breakthrough.Conventional conjugation methods are based on lysine and cysteine residues and the ADCs yielded by these methods have poor stability.They are prone to aggregation and the conjugated drugs are easy to shed resulting in non-therapeutic side effects.The ADCs produced by the site-specifc conjugation techniques developed recently have homogeneous drug-to-antibody ratios (DAR), similar pharmacokinetics with their parental antibodies and far less side effects than those of the conventional ADC with the same DAR.So the improved ADCs are expected to become a new generation of blockbuster drugs.Four site-specifc conjugation methods for ADCs were reviewed herein.
antibody-drug conjugate; site-specifc conjugation; reactive cysteine; unnatural amino acid; enzyme-based method; disulfde bond modifcation
R918
A
1001-5094(2015)04-0283-10
接受日期:2014-03-03
项目资助:国家自然科学基金(NO.81273425,81473125);“青蓝工程”江苏省中青年学术带头人培养对象项目
*通讯作者:张娟, 副教授;
研究方向:抗体药物的研究与开发;
Tel:025-83271483;E-mail:juancpu@126.com
