基于定点偶联技术的抗体药物偶联物的临床研究进展与挑战
2021-05-11陈虎张信玲孔娜娜罗文婷李乐乐刘丽娜黄长江姜静
陈虎,张信玲,孔娜娜,罗文婷,李乐乐,刘丽娜,黄长江*,姜静,3**
(1.荣昌生物制药(烟台)股份有限公司,山东 烟台 264006;2.烟台迈百瑞国际生物医药有限公司,山东 烟台 264006;3.滨州医学院,山东 烟台 264003)
1 抗体药物偶联物简介
抗体药物偶联物(ADC)由单克隆抗体(以下简称单抗)、高效细胞毒素及连接子组成。ADC 通过连接子将细胞毒素与抗体结合到一起,利用抗体对肿瘤细胞的生物特异性识别,直接把高效细胞毒素输送到肿瘤细胞,这种对肿瘤细胞的特异性识别提高了肿瘤部位高效细胞毒素的浓度,同时降低了高效细胞毒素在正常组织、器官中的暴露,从而达到增加抗肿瘤药效,降低对正常组织毒性的效果[1-3]。
然而,ADC的发展困难重重,经过近30年的发展,到2017 年之前,仅有2 个ADC 药物获得FDA 批准。由于ADC 研发成功率较低,其一开始并不被看好;但是,暂时的失败并没有让研究人员放弃对ADC 的深入探索。经过不懈努力,ADC 被证实可以达到良好的抗肿瘤疗效和安全性,现已成为抗肿瘤药物研发的新热点和重要趋势。截至2020 年12 月底,共有9 个ADC 获批用于肿瘤治疗(见表1),这对正在进行临床试验的80 多个在研ADC 是一个极大的鼓舞。

表1 已获批上市的ADC 一览Table 1 List of approved ADCs for the market
2 非定点偶联技术
ADC 的偶联方法主要包括非定点偶联和定点偶联。非定点偶联法是早期ADC 研究中使用的方法,是在不对抗体进行改造或修饰的情况下,利用化学方法直接将药物与抗体上氨基酸残基进行偶联,其偶联的细胞毒素个数和偶联位置都不能确定。非定点偶联包括赖氨酸偶联和半胱氨酸偶联。
抗体表面赖氨酸能和连接子上的琥珀酰亚胺(NHS)酯反应,构建ADC。IgG 分子具有超过80个赖氨酸残基,其中溶剂可及性高的位点超过20 个,而这些氨基酸均可被酰化,所以这些位点的偶联导致异质混合物的产生,因此采用赖氨酸偶联得到的ADC 异质性较强[4]。DAR 和细胞毒素的分布显著影响ADC 的药动学和药效学;此外,抗体可变区赖氨酸一旦被修饰,会影响抗体和抗原的亲和力。
抗体上的半胱氨酸和连接子上的马来酰亚胺(maleimide)发生反应,构建ADC。IgG 序列内的半胱氨酸残基较赖氨酸数量少得多,一个IgG 中有16 对二硫键,其中12 对为链内,4 对为链间。由于更高的溶剂可及性,4 对链间二硫键是偶联的潜在位点。这可以显着降低ADC 的异质性,以及获得比基于赖氨酸偶联的ADC 更高的均一性[5]。
总体来讲,非定点偶联ADC最终产物是混合物,包括了不一样的DAR 和不一样的偶联位点,其稳定性差,易发生聚集,且其中细胞毒素易脱落而产生非治疗性毒副作用,治疗窗较窄;此外,分析鉴定和控制生产批量之间的差异性也是技术上的大难题。为了克服这些难题,定点偶联技术随之出现。定点偶联技术通常需对抗体进行改造或修饰,能在特定位点实现细胞毒素的连接,提高了ADC 的均一性,这样不但便于分析鉴定和质量控制,还有利于改善药动学,增加ADC 治疗窗口。定点偶联将成为今后ADC 研发和创新的趋势[5-7]。
3 定点偶联技术
定点偶联技术大致可以分为5 类,图1 展示了这5 类技术的偶联位点:1)半胱氨酸偶联技术(图1 中“1”、“2”、“3”为偶联位点);2)工程化突变非天然氨基酸偶联技术(图1 中“4”为偶联位点);3)糖基偶联技术(图1 中“5”为偶联位点);4)氨基定点偶联技术(图1 中“6”为偶联位点);5)酶促多肽偶联技术(图1 中“7”、“8”、“9”为偶联位点)。

图1 定点偶联技术的偶联位点Figure 1 The combining site of site-specific conjugation techniques
3.1 半胱氨酸偶联技术
3.1.1 链间半胱氨酸高DAR 值偶联 2004 年,Seattle Genetics 公司报道了抗体链间半胱氨酸上连接2、4、8 个MMAE 的ADC(以下分别简称为D2、D4、D8)的有效性和安全性[8]。研究表明,尽管在体外细胞模型的评价中,D8 具有更强的细胞活性(D8 >D4 > D2),但其同等剂量下的体内抑瘤活性与D4基本一致;D2 在较高剂量下,能够保持与D4 和D8 相当的体内活性。D2 的最大耐受剂量分别是D4和D8 的2 倍和3 倍;同时,D8 的清除速率分别为D4 和D2 的3 倍和5 倍。综上所述,D4 的安全性和有效性综合评分较高,因而早期进入临床及获批的ADC 平均DAR 值也基本为4(3.5 ~ 4.5)。
虽然D8 具有较强的毒性和较快的清除速率,但其抗体中的8 个巯基全部与小分子反应,得到的ADC 具有确定的连接位置和确定的DAR 值,且不需要对抗体进行任何工程化改造,偶联工艺简便,是实现定点偶联的重要途径。
早期采用此策略的ADC(SGN-CD48A)主要使用含有聚乙二醇葡糖苷和MMAE 的连接子-毒素进行偶联。临床前研究表明此ADC 具有较好的抑瘤效果并进入Ⅰ期临床试验,然而,由于公司投资策略原因,终止后续开发。随后,第一三共公司和Immunomedics 公司分别开发出以低毒性喜树碱为毒素的连接子-毒素,即MC-GGFG-DXd 和CL2ASN-38。其中,第一三共公司的抗HER2 ADC,trastuzumab deruxtecan(Ds-8201a),于2019 年12月20 日获FDA 批准上市,用于无法切除或转移的HER2 阳性乳腺癌的治疗;Immunomedics 公司的sacituzumab govitecan(IMMU-132)于2020 年4 月被FDA 批准上市,用于已经接受至少2 种疗法的转移性三阴性乳腺癌的治疗。目前,还有labetuzumab govitecan(IMMU-130)、U3-1402 和SGN-CD228A等多款ADC 处于临床研究的不同阶段。
3.1.2 工程化突变半胱氨酸偶联技术 1)将抗体重链114 位的丙氨酸突变为半胱氨酸(HC-A114C)以及将轻链205 位缬氨酸突变为半胱氨酸(LC-V205C)。2008 年,基因泰克公司的Junutula 等[9]首次报道定点偶联技术能提升ADC 的治疗窗口。他们通过噬菌体展示技术,从赫赛汀(Heceptin)抗体轻链和重链中Fab 区域的恒定区中筛选适合定点突变的位点,最终确定将轻链110 位的缬氨酸突变为半胱氨酸(LC-V110C),以及HC-A114C 是定点偶联最佳的突变方式。随后,他们将突变的抗体与半胱氨酸反应性的探针结合,确定HC-A114C 抗体具有更好的偶联效率,并将此类定点偶联技术命名为THIOMAB。这项技术形成的抗体能够保持原始抗体的抗原亲和力和Fc 区域介导的生理活性,同时能够在不打开抗体链间二硫键的情况下,与巯基反应性连接子-毒素偶联,得到偶联位置确定、DAR均一的THIOMAB-药物偶联物(TDC)。这类药物既能够保持体内细胞活性,同时能够提高耐受性,降低全身毒副作用,提升治疗窗。目前,已有DMUC4064A、DCDS0780A 和BAT8003 这3 款 使用此类定点偶联技术的ADC 进入临床研究,其中BAT8003 是唯一一款仍处于临床活跃状态的产品。
2012 年,基因泰克公司的Shen 等[10]报道了第2 代THIOMAB 技术。研究表明,当突变位置具有较差的溶剂可及性,例如LC-V205C,其半胱氨酸具有带正电荷的环境,可以促进马来酰亚胺的水解,从而阻止逆迈克尔加成反应的发生,降低脱靶毒性。
2)将抗体重链239 位丝氨酸突变为半胱氨酸(HC-S239C)。Seattle Genetics 公司也开发了类似的技术[11-12]。他们选择HC-S239C,与以二聚吡咯开苯并吖庚三烯(PBD)为弹头的小分子定点偶联,开发了针对不同靶标的ADC,包括vadastuximab talirine、SGN-CD70A、SGN-CD352A、SGNCD19B、SGN-CD123A,ABBV-176 等多款ADC 药物,但遗憾的是,vadastuximab talirine 的Ⅲ期临床研究被宣告失败,至此所有临床上采用此突变方式的ADC 均宣告失败。
3)将抗体重链上442 位丝氨酸突变为半胱氨酸(HC-S442C)。ImmunoGen 开发的IMGN632,选择HC-S442C 为突变方式,目前该药处于Ⅰ/Ⅱ期临床活跃状态。
4)采用HC-S239C、HC-S442C 以及将重链上234 位亮氨酸突变为苯丙氨酸(HC-L234F)。阿斯利康研发的MEDI4276[13],选择对重链上的3 处位点进行突变,即HC-S239C、HC-S442C 及HCL234F。将靶向HER2 的双特异性抗体与定点偶联的4 个微管蛋白抑制剂相偶联。此ADC 的Ⅰ期临床试验被宣告失败。
5)将轻链183 位赖氨酸突变为半胱氨酸(LCK183C)以及将重链290 位赖氨酸突变为半胱氨酸(HC-K290C)。辉瑞开发的PF06804103 采用LCK183C 与HC-K290C 为突变方式,其避开轻重链的二硫键位点,达到定点偶联4 个auristatin 类的微管蛋白抑制剂,目前该药处于Ⅰ期临床试验活跃状态。
6)将重链220 位半胱氨酸突变为丝氨酸(HC-C220S)。 艾伯维(Abbvie) 开发的SC-003,采用HC-C220S 为突变方式,从而留下原本与重链220 位形成二硫键的轻链214 位半胱氨酸,与其自由巯基进行连接子-毒素偶联,使DAR 值达到2。此ADC 选择的毒素为PBD 类,其Ⅰ期临床试验被宣告失败。
7)将铰链区半胱氨酸突变为丝氨酸。ADC Therapeutics 开发的ADCT-502 与ADCT-602,以及阿斯利康开发的MEDI7247,均是将铰链区的4 个半胱氨酸突变为丝氨酸,剩下重链220 位的2 个半胱氨酸进行偶联而得到,这3 种ADC 均选择了PBD 毒素,目前ADCT-502 的Ⅰ期临床试验失败,其他2 种ADC 均处于Ⅰ期活跃状态[14]。
3.1.3 链间半胱氨酸桥接偶联技术 研究人员为能够在已经商业化的抗体上直接进行定点偶联,开发了桥接偶联技术。这项技术将抗体的4 对链间二硫键通过连接子进行桥接,重新将打开的二硫键连接起来,一方面可以得到DAR 值相对集中、偶联位置相对确定的ADC,另一方面能够保持链内二硫键连接,提升整体ADC 的稳定性。桥接偶联ADC 保留了抗原结合力,血浆稳定性好,且在体外和体内肿瘤模型中表现出较高效和具有良好抗原选择性的细胞杀伤活力。目前,基于双苯磺酸类桥接连接子与三丙烯基三嗪类桥接连接子这2 种桥接偶联连接子研发的ADC 已进入临床研究。
双苯磺酸类桥接连接子技术由PolyTherics/Abzena 公司开发[15]。其ADC 中DAR 值为4 的比例达到78%,没有裸抗体和高DAR 值(6 ~ 8)片段,均一度较高。2019 年8 月31 日,由OBI Pharma 公司研发的ADC 药物OBI-999,被美国FDA 批准进入Ⅰ期临床研究,用于治疗晚期实体肿瘤,随后,于同年12月26日获得孤儿药资格,用于治疗胰腺癌。
三丙烯基三嗪类桥接连接子技术由荣昌生物制药(烟台)有限公司开发[16]。其采用三丙烯酰基三嗪类连接单元,通过巯基与丙烯酰基的迈克尔加成反应,将IgG1 抗体的链间4 对二硫键桥接,得到DAR 值为4 的比例超过70%的ADC 分子。此类连接子可以在抗DR5 的抗体中引入负载一甲基澳瑞他汀D(MMAD)的连接子-毒素,并在体外和体内对白血病和实体肿瘤具有很好的抗肿瘤增殖活性[17]。2018 年4 月,由荣昌生物制药(烟台)有限公司开发的RC88 ADC 获得临床批件。目前,正在进行RC88 项目的Ⅰ期临床试验。
3.2 工程化突变非天然氨基酸偶联技术
该技术通过在抗体的原始序列中人为加入非天然氨基酸,使其在抗体表面呈现可方便偶联的特异性位点,连接子-毒素与特定位点相偶联,从而达到定点偶联的目的。
3.2.1 Xpress CF+技术 Sutro Biopharma 开发了Xpress CF+技术,研发了Stro-001 与Stro-002,目前这2 个ADC 处于Ⅰ期临床试验活跃状态[18]。
Stro-001 在每个重链上的F404 位置都含有2 个对叠氮苯丙氨酸(pAMF)残基,定点偶联2个连接子-毒素,其采用了不可裂解的二苯并环辛(DBCO)-美登素作为连接子-毒素。
Stro-002 在每个重链上的Y180 和F404 位置都含有4 个pAMF 残基,从而达到定点偶联4 个连接子-毒素的目的。其采用一种新型的连接子-毒素SC239,SC239 由以微管蛋白为靶标的3-氨基苯基半胱氨酸弹头(SC209)和用DBCO 功能化的一种可裂解的缬氨酸-瓜氨酸对氨基苄酯连接子构成。DBCO 对pAMF 残基的快速选择性反应保证了ADC的均一性,使DAR 值保持在4。
3.2.2 EuCODE 技术 Ambrx 开发的ARX788[19-20]是通过EuCODE 技术在抗体一级序列的特定位点掺入非天然的对乙酰苯丙氨酸(pAcF)而得到。这种非天然氨基酸通过稳定的肟键为多种有效载荷的共价结合提供了一个可链接的平台。ARX788 选择不可裂解的连接子与MMAF 进行偶联,平均DAR 值达2。目前此ADC 处于Ⅰ期临床试验活跃状态。
Astellas 与Ambrx 共同开发的AGS62P1[21-22]同样是插入非天然氨基酸,在其抗体每个重链的124位点嵌入pAF 残基,抗体通过pAF 位点上的肟键与有效的细胞毒素MMAF 的衍生物AGD-0182 进行偶联,使平均DAR 值达到2。目前此ADC 处于Ⅰ期临床试验活跃状态。
3.3 糖基偶联技术
天然抗体上具有不同类型的糖基修饰,这些不同的糖基可被糖苷内切酶修饰,从而曝露出N-乙酰氨基葡萄糖,经叠氮修饰后的N-乙酰半乳糖胺利用糖基转移酶连接到抗体的N-乙酰氨基葡萄糖上。被叠氮修饰后的抗体进一步与双环酮修饰的连接子-毒素发生点击化学反应[23],得到定点偶连的ADC。此类技术由Synaffix 公司开发,称为GlycoConnect技术。
ADCT-601(BGB601) 由 Synaffix 的GlycoConnect 技术平台开发。ADCT-601 靶向受体酪氨酸激酶AXL,其在许多实体瘤和血液系统恶性肿瘤中高度表达[24]。ADCT-601 通过可裂解的缬氨酸-丙氨酸连接子将人源化IgG1 抗体和细胞毒素PBD连接起来。在临床前试验中,ADCT-601 在具有不同AXL 表达水平的各种肿瘤异种移植模型中均有效,目前该ADC 处于Ⅰ期临床试验阶段。
3.4 特定赖氨酸的氨基定点偶联技术
早期开发的ADC 药物, 例如Mylotarg、Kadcyla 和Besponsa,均利用抗体上的赖氨酸连接小分子药物。然而,由于抗体上赖氨酸数量众多、化学性质相似,导致偶联的位置和数量高度不均一。肽图研究表明,抗体上能够发生偶联的赖氨酸约有40 个,因此形成的ADC 结构可高达百万种。这给药物的生产工艺控制和检测造成了极大困难,也影响了药物的有效性和安全性[25]。
目前,对氨基进行定点偶联的主要策略是,依赖赖氨酸在特定pH 值下与亲核试剂的反应速率差异,实现定点偶联。由于赖氨酸的位置和微环境的差异性,质子化的胺基的pKa 各不相同,其与亲核试剂的反应速率有显著差异。利用此性质差异,选择适当的连接头,可以选择性地选择特定赖氨酸残基用于偶联[25]。
2017 年7 月,由四川科伦公司开发的ADC 药物——A166 在我国获批进入Ⅰ期临床,用于HER2阳性的乳腺癌治疗。A166 是全球首个应用氨基定点偶联技术制备形成的ADC 药物。已公开的专利表明[26],通过创新性的连接子设计,将小分子药物定点连接到赫赛汀抗体轻链上的特定序列。2018 年,此ADC 也获得FDA 的Ⅰ期临床试验许可。
3.5 多肽酶促偶联技术
3.5.1 SMARTag 技 术 Redwood Bioscience 开 发 了SMARTag 特定位点的蛋白质修饰和接头技术。该技术通过在抗体特定位置引入CxPxR 序列,该序列中的半胱氨酸通过甲酰甘氨酸生成酶,翻译后识别、修饰产生醛,而后有效载荷通过Hydrazino-Pictet-Spengler(HIPS)反应与醛形成稳定偶联物。SMARTag 平台可提供精确的有效载荷,针对特定位置的结合。
由Triphase Accelerator 与Celgene 合作开发的TRPH-222,是基于此技术的ADC。其药物与抗体的平均DAR 为1.8,目前处于Ⅰ期临床活跃状态。其有效负载位置、优化的接头组成与HIPS 化学提供的稳定性相结合,可带来更好的耐受性和扩展的治疗指数[27]。
3.5.2 ConjuAll 技 术 ConjuAll 技 术 是 将CAAX、XXCC、XCXC 或CXX 序列(C 代表半胱氨酸,A代表脂肪族氨基酸,X 代表决定类异戊二烯转移酶的底物特异性的氨基酸)修饰到抗体的C 端。该序列可被类异戊二烯转移酶(FTase 或GGTase)特异性识别,将含有荧光团、生物素、炔、叠氮的功能性官能团上的异戊二烯与该序列半胱氨酸的巯基相偶联,最后通过化学反应将连接子-毒素连接起来[28]。由LegoChem Biosciences 与复星医药合作开发的FS-1502(LCB14-0110)采用的就是LegoChem 公司的ConjuAll ™技术,目前处于Ⅰ期临床活跃状态。
3.5.3 NexMab 技术 NexMab 技术系在抗体上引入含有半胱氨酸的多肽,通过金属离子保护游离的半胱氨酸,从而选择性偶联不受保护的半胱氨酸,实现定点偶联。在偶联过程中,C 端引入的含有半胱氨酸的肽链可以通过金属离子的配位作用,阻止半胱氨酸偶联。由Alteogen 基于NexMab 技术开发的ALT-P7 产品,是以乳腺癌、胃癌为适应证的ADC。由于引入定点偶联技术,ALT-P7 或可被开发为比Kadcyla 更安全、更有效的抗癌药物[29]。目前正在进行该药的Ⅰ期临床试验。
基于定点偶联技术的ADC 如表2 所示。
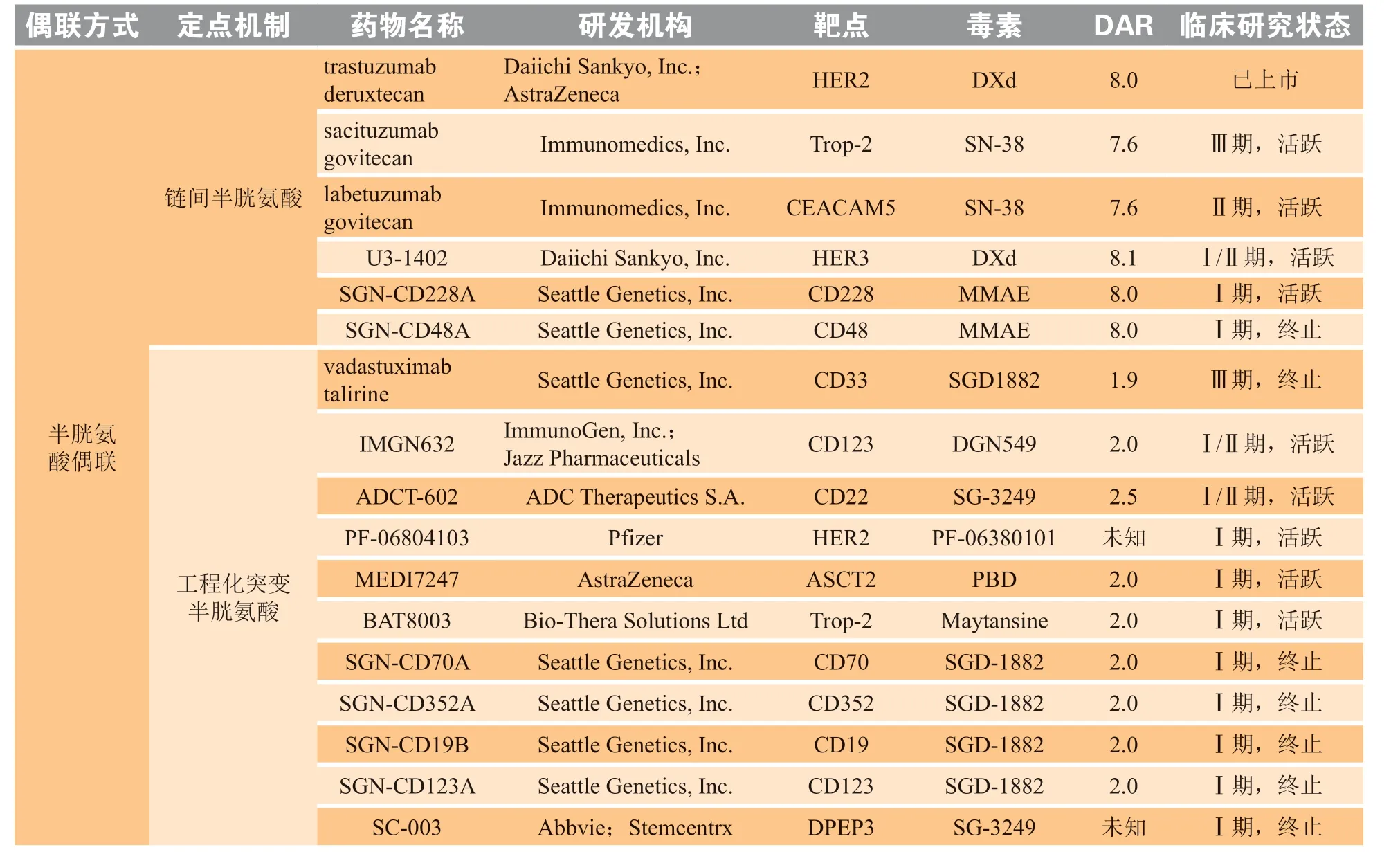
表2 基于定点偶联技术的抗体药物偶联物一览Table 2 List of clinical ADCs based on site-specific conjugation techniques

续表2
4 定点偶联ADC 的临床疗效
定点偶联的ADC 除了DS-8201a(trastuzumab deruxtecan)和IMMU-132(sacituzumab govitecan)以外,均处于研究初期(Ⅰ期临床阶段),披露的临床数据较少(见表3)。
DS-8201a 是一种将4 对链间二硫键全部还原后偶联的ADC,DAR 为8 左右,连接子为马来酰亚胺。DS-8201a 采用的GGFG 连接子,具有很好的稳定性。据报道,药动学(PK)实验中发现,DS-8201a和总抗的血浆浓度之间存在很小的差异,而T-DM1(Kadcyla 的研究代码)在猴PK 研究中被发现与总抗之间的浓度存在明显差异。此外,血浆DXd 浓度明显低于血浆DS-8201a 浓度,且DS-8201a ADC 和细胞毒素之间的暴露差异比T-DM1 的相应差异大1个数量级。这些结果表明DS-8201a 的接头是稳定的。这与以下事实相符:即使将DS-8201a 在食蟹猴血浆中体外培养21 d 后,也仅释放约4%的DXd,而T-DM1 96 h 时就有将近20%的有效载荷从T-DM1释放。因此,即使在DAR 7 ~ 8,DS-8201a 的药物接头也比T-DM1 更稳定(平均DAR 约为3.5)。另外,对猴子静脉注射DXd 后,DXd 从体内迅速清除,清除率接近猴子的肝流速。也就是说,除了接头稳定性之外,DXd 的高清除率也是DXd 暴露量低的主要原因之一[30]。
DS-8201a 的精妙设计在临床上也显示了出色的临床结果。最经典的临床试验是DESTINYBreast01,实验结果一经公布,ADC 业界为之振奋。DESTINY-Breast01 研究是一项全球多中心、开放标签的Ⅱ期临床研究,纳入T-DM1 耐药/难治性的HER2 阳性不可切除和(或)转移性乳腺癌患者,探索了DS-8201a 治疗的有效性与安全性。研究共2个部分:第1 部分纳入65 例患者,按1∶1∶1 随机分为3 组(低、中、高剂量),进行PK 评估,在此基础上,再纳入54 例患者,按1∶1 随机接受低剂量(5.4 mg · kg-1)和高剂量(确定的推荐剂量:6.4 mg · kg-1)的DS-8201a 治疗。第2 部分为开放标签的持续阶段,约纳入134 例患者,接受DS-8201a推荐剂量(5.4 mg · kg-1)治疗。另外,约有4 例TDM-1 不耐受患者将作为探索性分支进入持续研究阶段。表3 的临床结果展现了DS-8201a 良好的有效性和安全性[31]。
其实在DESTINY-Breast01 这项Ⅱ期研究之前,针对既往使用T-DM1 治疗的HER2 阳性乳腺癌患者的一项Ⅰ期研究(DS8201-A-J101)已经让DS-8201a 显示出积极的抗肿瘤活性。结果显示,在既往平均接受过7 种抗肿瘤疗法的入组人群中,中位治疗线数不低于7 线的情况下,DS-8201a 治疗后患者仍可获得59.5%的客观缓解率(ORR),疾病控制率(DCR)为93.7%,缓解持续时间(DOR)为20.7 个月,无进展生存期(PFS)达22.1 个月,且尚未达到中位总生存期(mOS)[32]。该项Ⅰ期研究同时披露了DS-8201a 治疗其他肿瘤的相关结果[32-35]。在胃癌中,DS-8201a 的表现略逊于对乳腺癌的疗效,ORR 达43.2%,DCR 达79.5%,中位DOR 为7 个月,中位PFS 为5.6 个月,mOS 为12.8 个月,但这在同类型的药物中也算是比较突出的结果。在非小细胞肺癌(NSCLC)中,ORR 达58.8%,DCR 达88.2%,中位PFS 达14.1 个月,中位DOR 达9.9 个月,但入组人数少,结果可能偏差较大。在结直肠癌中,效果较差,ORR 仅为15.8%,DCR 可达84.2%。在安全性方面,该药对不同适应证结果相似(见表3)。
2019 年的圣安东尼奥乳腺癌研讨会(SABCS)上,一项Ⅰ期临床研究结果被公布(见表3)。该试验与其他试验不同之处在于,入组病人中92%为HER2 低表达,其中IHC 1+占73%,IHC 2+/ISH-占12%。试验结果表明,ORR 达39.0%,DCR 达84.0%,说明DS-8201a 对HER2 低表达的病人也有较好疗效[36]。NCT03383692 是一项将DS-8201a和利托那韦以及伊曲康唑联用的试验,ORR 为41.70%,表现良好[37]。
IMMU-132 是另一款已上市的定点偶联ADC,其定点偶连原理与DS-8201a 相似。但是IMMU-132连接子设计理念与传统ADC 不同,传统ADC 的连接子在血清中保持相对较高的稳定性,而在IMMU-132 的研究过程中发现,SN-38 与各种具有不同释放速率的接头偶联制备的ADC 中,具有中等释放速率的ADC 展现出最佳的治疗活性[38]。IMMU-132对尿路上皮癌和三阴性乳腺癌这2 种适应证表现较好(见表3)[39-40]。IMMU-132 是第1 款治疗三阴性乳腺癌的ADC,对于只能化疗的三阴性乳腺癌患者,IMMU-132 提供了一种理想的靶向治疗手段[40]。
U3-1402 是一种HER3 靶向ADC,在治疗转移性表皮生长因子受体突变且对酪氨酸激酶耐药的NSCLC 患者的临床研究中,最新研究结果报告了U3-1402 的有效性,在26 名可评价的患者中,22 名患者在所有剂量下都有6 个已确认的部分缓解(PR),且在具有不同耐药机制的患者中观察到PR 和肿瘤缩小[41]。该药对乳腺癌疗效优于对NSCLC,ORR达42.9%,DCR 达90.5%[42]。IMGN632 用于治疗急性髓系白血病(AML),ORR 仅为13%,药效欠佳[43]。ARX788 使用的是非天然氨基酸定点偶联技术,最近的报道表明ARX788 对乳腺癌疗效较好,ORR 高达63%,且未发现剂量限制性毒性[44]。
5 定点偶联ADC 的挑战及发展方向
目前失败的ADC 有91 个,其中定点偶联ADC占14 个。在这终止研究的14 个定点偶联ADC中,有9 个使用的是毒素PBD,其中vadastuximab talirine 进展到了Ⅲ期临床。2017 年,基因泰克宣布Ⅲ期临床试验CASCADE 终止。CASCADE 获得的数据表明,在接受vadastuximab talirine 治疗后,患者死亡率更高。由此,基因泰克暂停了vadastuximab talirine 临床试验受试者的招募,同时也停止了该药正在进行中的临床试验。与已上市的同靶点(CD33)ADC 药物gemtuzumab ozogamicin相比,vadastuximab talirine 使用了新一代的PBD 弹头,细胞内酶促裂解的缬氨酸-丙氨酸二肽连接头作为释放装置,定点突变的半胱氨酸与马来酰亚胺的迈克尔加成反应作为连接技术。Vadastuximab talirine 的主要失败原因是其毒性反应强烈,患者死亡率高,这可能是由于PBD 毒素的高细胞毒性,在循环系统中意外释放,从而引起显著的全身毒性。MEDI4276 由于有效性低而被终止研究,治疗HER2 阳性乳腺癌的临床试验表明其ORR 仅为4%,这可能与该药特殊的双表位抗体有关系。DMUC4064A 的一项临床试验显示ORR为45%,安全性也能达标,终止原因未知[45]。
2019 年, 由罗氏/基因泰克公司开发的polatuzumab vedotin 被批准上市,与苯达莫司汀和rituximab 联合使用,用于治疗难治的弥漫性大B 细胞淋巴瘤(DLBCL)成人患者。在此药物的早期临床阶段,有一款同靶点、同抗体、同小分子的ADC(DCDS0780A)也进入了Ⅰ期临床研究。在一项针对B 细胞非霍奇金淋巴瘤的Ⅰ期临床研究中,DCDS0780A 与rituximab 和obinutuzumab 联 用组的ORR 为40.00%,其中完全缓解(CR)率为14%(6/43);PR 率为26.00%(11/43),但并未公开终止原因[46]。相比之下,DCDS0780A 的CR(14%)显著低于Polivy(40%)。Polatuzumab vedotin 使用非定点偶联技术,其平均DAR 值为3.5,而DCDS0780A 使用定点偶联技术,其DAR 值为1.9。可以推测,载药量偏低引起的有效性不足可能是DCDS0780A 未能进一步进入临床研究的重要原因。
定点偶联ADC 的临床成功率并不高,其临床试验失败的原因很多,有可能是毒素不合适、DAR偏低,有可能是靶标不好,也有可能是连接子不稳定,或者是公司决策的原因。因此,定点偶联的低成功率不能说明定点偶联的方式不好。相反,定点偶联仍是以后ADC 发展的方向,毕竟定点偶联可以解决传统ADC 组成复杂的问题。最近上市的DS-8201a 和IMMU-132 均是基于定点偶联技术研制,这给定点偶联ADC 的成功照亮了道路。

表3 基于定点偶联技术的抗体药物偶联物的临床数据Table 3 Clinical data of ADCs based on site-specific conjugation techniques

续表3

续表3
