基于设计的生物大分子成药性优化策略研究进展
2015-02-10田浤尹俊戴鑫宁宏宇陈松高向东姚文兵
田浤,尹俊,戴鑫,宁宏宇,陈松,高向东,姚文兵
(中国药科大学生命科学与技术学院, 江苏 南京 210009)
基于设计的生物大分子成药性优化策略研究进展
田浤,尹俊,戴鑫,宁宏宇,陈松,高向东,姚文兵*
(中国药科大学生命科学与技术学院, 江苏 南京 210009)
目前生物药物正处在高速发展阶段,但生物大分子的一些固有特性限制了其成药性,使得很多具有良好治疗潜能的生物大分子最终不能开发成药物,因而严重制约了生物药物的发展。生物药物开发的瓶颈已从“新分子的产生”转向“如何获得具有优良生理特性和预期治疗效果的有效药物”。近年来,通过合理设计改造生物大分子高级结构以优化其成药性的研究获得了快速发展。综述基于设计的生物大分子成药性优化策略研究进展。
生物大分子;成药性;合理设计
随着基因组学、蛋白质组学等学科的发展,被发现具有特定活性的生物大分子数量每年呈指数增加,但最终成为药物而应用于临床的生物大分子数量却很有限,其中一个重要原因就是生物大分子的一些固有特性限制了其成药性。生物大分子具有结构复杂、易产生免疫原性、稳定性差、半衰期短、多数需频繁给药、生物利用度低、依从性差等特性,这些特殊性使很多具有良好生物活性的生物大分子最终不能开发成药物,严重制约了生物技术药物的发展。2003—2011年的统计分析显示,9年中美国FDA共批准了1 173 个生物药物及其对应的适应证进入临床研究阶段,但最终获批用于临床治疗的仅171个药物及其对应的适应证。生物药物开发的瓶颈已经从“新分子的产生”转向“如何获得具有优良生理特性和预期治疗效果的有效药物”,生物药物成药性研究已成为国内外生物医药产业界共同关注的热点问题。
与小分子化学药物相比,生物大分子的结构更为复杂,且其高级结构与其生物活性、稳定性、免疫原性等性质密切相关,而这些性质往往又是影响生物大分子成药性的关键因素。近年来,随着生物信息学、结构生物学和各种组学技术的发展,通过合理设计改造生物大分子高级结构以优化其成药性的研究获得了快速发展。本文就基于设计的生物大分子成药性优化策略研究进展作一综述。
1 生物大分子药代动力学性质的优化策略
生物大分子的成药性受到多种因素的影响和制约,其中,药代动力学性质与药物治疗效果及安全性都高度相关,是最为关键的因素之一。很多具有良好生物
活性的候选生物大分子由于药代动力学性质不佳而无法应用于临床,故药代动力学性质的优化一直是生物大分子药物开发中的一个研究热点。
1.1 增加生物大分子流体力学半径
对于相对分子质量小于60 000的生物药物分子,肾小球滤过是其主要的消除途径。因此,增加分子的流体力学半径,减少肾小球滤过是优化其药代动力学性质的有效策略之一[1]。
增加分子的流体力学半径最成熟的技术是聚乙二醇化技术(PEGylation),目前已用于多种蛋白质药物或非蛋白质药物的修饰,国外上市的PEG化药物已达11种[2]。虽然PEG修饰已经成为现在最为成熟、应用最为广泛的生物药物长效化技术之一,但其也存在着生产工艺复杂、产物不均一、肾脏中易聚集等问题。
为了解决这些问题,近年来国内外有研究人员开发出模拟PEG的聚多肽融合技术(recombinant polypeptide mimetics of PEG)。其原理是,通过亲水、柔性、无固定二级结构的多肽链替代PEG用于生物药物修饰,以提高药物的流体力学半径,减少其肾小球滤过。聚多肽融合技术的核心环节是聚多肽链的设计,早期研究人员大多采用天然来源的聚多肽、明胶样蛋白聚多肽、弹性蛋白样聚多肽、多聚谷氨酸、多聚甘氨酸等聚多肽,但这些聚多肽多存在着免疫原性高、易聚集、不能显著增加半衰期等问题[3-5]。美国Amunix公司开发了一种采用全人工设计的低免疫原性短肽非重复组合而形成的聚多肽链XTEN,该聚多肽链只含有Ala、Glu、Gly、Pro、Ser、Thr等6种氨基酸,具有可生物降解、无毒性代谢过程、无肾脏堆积风险等优势(见图1)[5]。

图1 聚多肽的修饰通过增加生物药物的流体力学半径而减少药物肾小球滤过Figure 1 Polypeptide modification results in the increase of hydrodynamic radius of biological drug and the decrease of its glomerular filtration
XTEN与艾塞那肽(exenatide)融合表达后,使艾塞那肽在人体中半衰期从原来的2.4 h延长至139 h。类似的,XTEN修饰的重组人生长激素(rhGH)VRS-317在高剂量下的终末半衰期能达到131 h,相比于每天注射的未修饰rhGH,其能更加高效、持续地刺激胰岛素样生长因子-Ⅰ(IGF-I)应答,目前VRS-317已进入临床Ⅱ期研究[6]。眼下,采用XTEN技术进行药代动力学优化的生物大分子还包括胰高血糖素、胰岛素、凝血因子Ⅶa、凝血因子Ⅷ、凝血因子Ⅸ等。
1.2 优化受体介导的再循环过程
受体介导的胞吞作用也是生物药物从血液中消除的主要途径之一,而生物大分子通过受体胞吞进入细胞后的转运途径是影响其血浆半衰期的关键因素之一。一些生物大分子能通过内涵体再循环过程回到细胞表面,重新释放到血液循环中,如:免疫球蛋白G(IgG)抗体在早期酸化的内涵体中与Fc受体(FcRn)结合,从而逃离了被胞内溶酶体降解的命运,被再次释放进入血液。这是IgG抗体类药物具有较长血浆半衰期的主要原因。因此,增加受体介导的再循环过程是优化生物大分子药代动力学性质的重要策略。
借助受体介导再循环过程延长药物血浆半衰期的Fc融合技术与白蛋白融合技术已发展得较为成熟,本
文不再赘述。近年来,这一策略的主要研究进展集中在对与FcRn具有结合能力的融合伴侣的设计与改造。
受体介导的再循环过程具有明显的pH依赖性。IgG与FcRn在酸性条件下结合,生理环境中解离。所以,通过对IgG的合理设计与改造,增强其与FcRn在酸性条件下的亲和力,而不增加它们在生理环境中的亲和力,则可以有效增加FcRn介导的再循环过程。IgG上与FcRn识别的位点位于IgG Fc片段的CH2和CH3结构域,Zalevsky等[7]通过将IgG Fc片段中的428位Met突变成Leu及434位Asp突变成Ser,使IgG分子在pH 6条件下与FcRn的亲和力提高11倍。随后该研究团队将这项被称为Fc engineering的技术应用于单抗药物bevacizumab和cetuximab的修饰改造,以增加药物的血浆半衰期。实验研究显示,Fc engineering技术的应用使bevacizumab在猕猴体内的半衰期从9.7 d增加到31.1 d,cetuximab的血浆半衰期也从2.9 d延长至13.9 d。
融合人血白蛋白(HSA)也是通过FcRn受体介导的再循环过程而延长药物血浆半衰期的主要技术之一。随着HSA与FcRn的结合方式在分子水平上的进一步阐明,人们发现HSA的第3结构域(DIII)在HSA与FcRn的结合过程中起主要作用[8]。据此,Kenanova等[9]选用HSA的DIII为融合伴侣,将其与一个抗癌胚抗原的双抗体结合,所得融合蛋白不仅保持了原抗体的肿瘤靶向性,且其血浆清除率明显降低,血浆滞留时间得到了大幅度延长。
2 生物大分子药效学性质的优化策略
2.1 优化生物大分子与靶受体的亲和力
以蛋白质为主的生物大分子类药物大多是通过与靶受体结合而发挥作用,故对受体亲和力的优化是改善生物大分子药效学性质的主要策略之一。通常,在保持活性中心的情况下,增加与靶蛋白结合的亲和力,可以提高生物大分子的生物活性;而在不改变亲和力的情况下,通过突变使生物大分子失去活性中心,则可以开发出对应的拮抗剂。
瘦素是一种多效激素,在能量代谢、生殖和免疫等生物学过程中起着重要的调节作用,并且也是最重要的体质量调节因子。Shpilman等[10]对瘦素进行随机突变,并用酵母表面展示技术筛选出高亲和力的突变。结果发现,当瘦素第23位的Asp突变为非负电荷性的氨基酸时(即 D23L),能极大增强瘦素与其可溶型受体的亲和力,而第39、40、41位氨基酸同时突变为Ala时(即L39A/D40A/F41A),突变体则表现出瘦素拮抗剂活性。体外实验结果表明,这种新人源(SHLA)和鼠源(SMLA)瘦素拮抗剂(D23L/L39A/D40A/F41A)的受体亲和力和拮抗活性分别较L39A/D40A/F41A提高了60倍和14倍。
2.2 提高生物大分子的作用靶点选择性
具有良好的作用靶点选择性是生物大分子成药的一个关键环节。药物与非期望的靶点发生作用往往会带来诸多副作用,所以通过合理设计而赋予生物大分子对目标靶点的选择性,也是其成药性优化的关键策略。
白介素-2(IL-2)是免疫系统的一个重要调控因子,可刺激T细胞增殖,对多种肿瘤的生长有强抑制作用。然而由于IL-2会引起一些严重的毒副作用,如引起肺液聚集导致肺水肿,最终引发呼吸困难,使其临床应用受到限制。最近,斯坦福大学的Levin等[11]通过体外进化,设计了一种IL-2超级因子super-2,增强了IL-2对其受体IL-2Rβ的亲和力,从而消除了CD25表达对IL-2的依赖性。游离型和结合型super-2的晶体结构显示其进化突变位点位于该细胞因子的核心,且分子动力学模拟提示该突变能提高IL-2的稳定性,减少一个位于IL-2Rβ结合位点的螺旋的灵活性,将IL-2优化为一种类似于与CD25结合时的受体结合构象。super-2中的进化突变替代了CD25的作用,可引发STAT5的有效磷酸化,并使T细胞不论CD25是否存在都能旺盛增殖。相对于IL-2,super-2能更强地介导细胞毒性T细胞扩增,在体内介导更强的抗肿瘤反应,并引起调节性T细胞比例降低,减轻肺水肿症状。
3 生物大分子免疫学性质的优化策略
3.1 降低生物大分子免疫原性
细胞因子、酶、抗体等生物大分子易产生有害的免疫反应,这是制约其安全性的一个重要因素,也是生物药物成药性研究中值得关注的一个热点问题。消除生物大分子免疫原性的最普遍的方法是通过合理置换抗原表位的氨基酸残基,使所产生的序列对已知中和抗
体具有低亲和性。徐瑞光等[12]为了降低葡激酶的免疫原性,尝试对其T/B细胞抗原表位重叠的77位Arg和80位Glu进行突变。实验结果表明,当80位Glu突变为Ala和Ser时,能同时去除部分T/B细胞抗原表位;当77位Arg突变为Asn、Gln和Lys时,仅去除了部分T细胞表位;在6个突变组合中,发生R77Q/E80A突变和R77Q/E80S突变的葡激酶有效去除了部分B/T细胞抗原表位,降低了葡激酶的免疫原性,而发生R77Q/ E80A突变和R77Q/E80S突变的葡激酶其酶活力与未突变前相当。
另一种降低生物大分子免疫原性的策略是致使与Ⅱ型主要组织相容性复合体(MHCII)类分子结合的肽段突变而阻止T细胞活化,而除去与MHC分子结合的抗原表位,是降低蛋白质类药物免疫原性的一种更为温和的方法,优于除去抗体上抗原表位的方法,因为MHC分子仅有1×103~2×103种突变体,而抗体总体估计大约有108种突变体。Salvat等[13]设计了一种基于分析蛋白质结构和免疫原性从而对氨基酸位点进行突变的计算方法,并利用此算法设计了7个候选的β-内酰胺酶药物,这些药物均大大减少了与人MHCII蛋白结合的表位,因而免疫原性降低,且均保持了良好的稳定性和活性。
3.2 提高生物大分子免疫原性
目前,治疗性疫苗在肿瘤和自身免疫性疾病治疗等领域的应用日益受到重视,而许多重要的治疗靶点,如肿瘤相关抗原MAGE、gp100等,都是自体蛋白,因而存在自身免疫耐受而无法直接用于疫苗的开发。因此,近年来研究人员提出许多提高目标分子免疫原性或突破自身免疫耐受的优化策略。
将免疫原性氨基酸引入蛋白质分子中,即是突破自身免疫耐受的一种新技术(见图2)。借助近年来在分子生物学领域迅速发展起来的“遗传密码扩充技术”,Grü newald等[14-15]成功将对硝基苯丙氨酸(pNO2Phe)等免疫原性氨基酸定点引入来源于小鼠的肿瘤坏死因子-α(TNF-α)和视黄醇结合蛋白4(RBP4)等蛋白中,并用于激活小鼠的体液免疫应答,引起了高滴度IgG抗体的产生,打破了自身免疫耐受。Gauba等[16]同样运用遗传密码扩充技术将pNO2Phe、3NO2Tyr或SO3Tyr定点引入TNF-α和表皮生长因子(EGF)蛋白中,并证明了免疫原性氨基酸是通过激活CD4+T淋巴细胞而激活小鼠的体液免疫应答,而其是否能打破免疫耐受与突变的位点和小鼠的MHCII类分子相关。Hardy等[17]将抗原表位肽中的Tyr置换成NO2Tyr,并证实,仅在一个位点引入免疫原性氨基酸,即可明显影响MHCI类限制性抗原表位的识别。
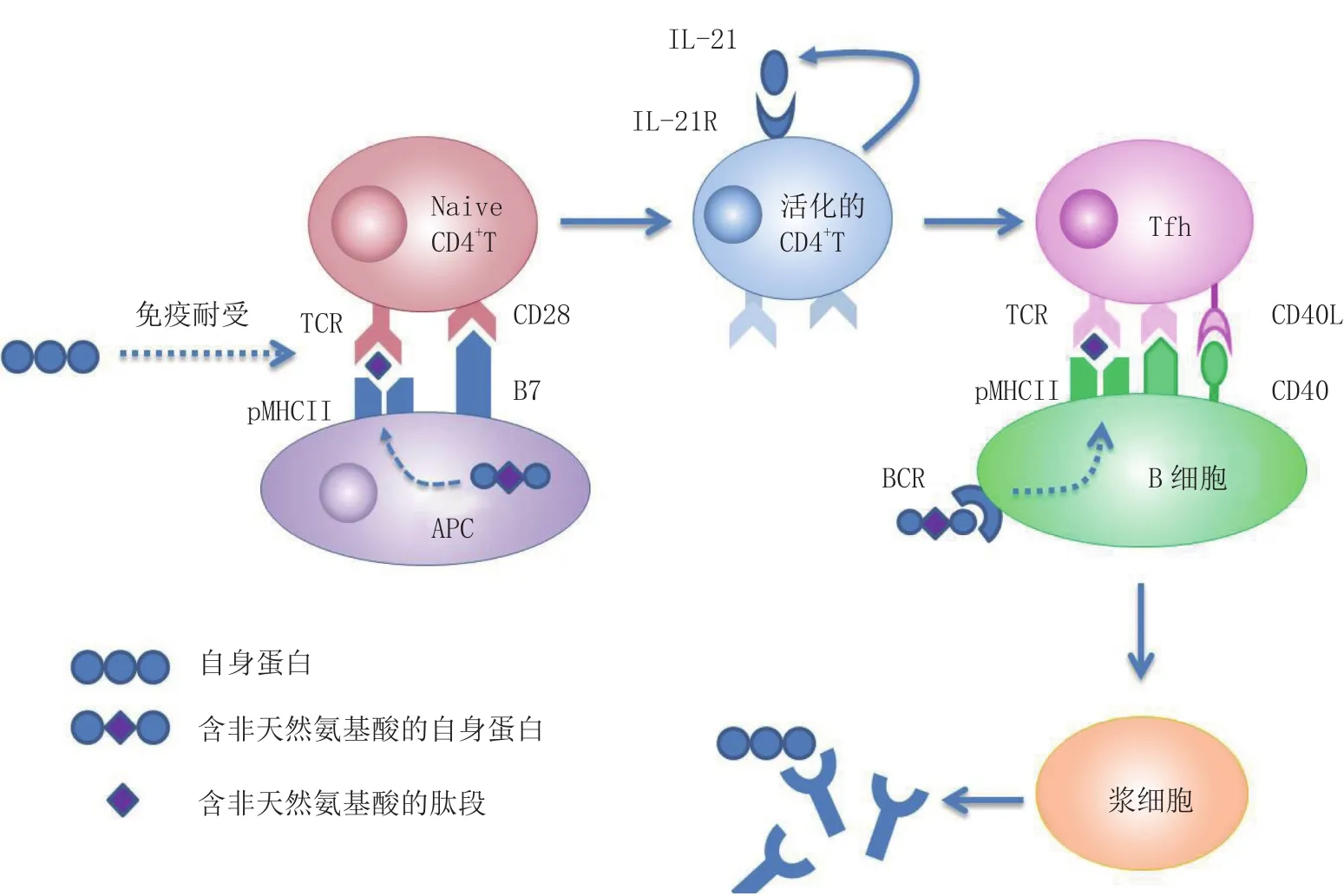
图2 通过引入非天然的免疫原性氨基酸致使蛋白质突破自身免疫耐受的策略Figure 2 Strategy for breaking self-tolerance of protein based on incorporating unnatural immunogenic amino acid
另一种提高生物大分子免疫原性的策略是将目标分子靶向到特定淋巴细胞亚群,通过交叉递呈的方式提高机体免疫应答功能。Klechevsky等[18]尝试将肿瘤抗原通过抗树突细胞免疫受体(DCIR)抗体靶向到DCIR受体上,成功促进了树突细胞的交叉递呈功能,强烈激活了CD8+T细胞。Idoyaga等[19]将HIV gag p24蛋白靶向到Langerin、DEC205和Clec9A受体上后,都能不同程度地引起小鼠细胞免疫和体液免疫应答。
4 生物大分子理化性质的优化策略
生物大分子的理化性质对其成药性也有着重要影响。具有研发价值的生物大分子往往要求具有良好的溶解度,且能在纯化、制备、贮存和给药过程中保持稳定,但许多具有治疗潜能的天然生物大分子往往不能满足这种要求。因此,对生物大分子理化性质进行优化,也是成药性优化策略的重要环节,如提高生物大分子的抗氧化能力、pH和温度稳定性以及溶解性,以利于其发挥药效和扩大使用范围。
分布在蛋白质表面的带电荷氨基酸之间可以通过电荷间的相互作用而形成保护网,增加蛋白质对高温的抗性。因此,可以通过对蛋白质表面带电荷的氨基酸进行重新设计优化,使其表面的电荷分布更加合理,从而提高其稳定性。藉此,Gribenko等[20]通过软件分析对酰基磷酸化酶和GTP酶进行了重新设计,结果发现,这些改进型酶的热稳定性较野生型酶提高了10 ℃。
根据蛋白质中二硫键键长和键角的信息,分析蛋白质中哪些位点可以形成二硫键,并通过突变引入Cys,形成二硫键,这样可明显提高蛋白质的稳定性[21]。Mateo等[22]通过在手足口病病毒衣壳蛋白中合理引入二硫键,使其热稳定性大幅度提高而感染活性消失,为后续疫苗的开发奠定了基础。
另一方面,在蛋白质药物中置换不成对的Cys,能防止不期望的分子间二硫键的形成。游离的Cys易在蛋白质分子内部和分子之间形成二硫键,致使分子呈聚集状态,影响药物的可溶性和稳定性,降低其生物利用度。将蛋白质药物中Cys突变为Ser,可防止其二硫键的形成,从而改善药物的溶解度和贮藏稳定性。Chiron公司生产的Proleukin和Berlex/Chiron公司生产的Betaseron等蛋白质药物就是通过将游离Cys突变而成功改善其理化性质。
5 结语
随着现代生物技术的发展,对于严重威胁人类健康的重大疾病,如遗传性疾病、癌症、糖尿病等的治疗,生物药物的作用已变得举足轻重,甚至不可替代。
生物信息学、结构生物学和各种组学技术的快速发展促进了人们对生物药物的空间结构、作用靶点、细胞转运途径等的理解和认识,为理性设计生物大分子药物提供了理论和技术支持,也使得通过合理设计改造生物大分子的高级结构以优化其成药性的研究获得了快速发展。可以相信,随着生命科学的进一步发展以及人们对生命活动理解的进一步加深,会有越来越多的活性生物大分子通过合理的成药性优化而进入临床,为保护人类健康发挥重要作用。
[1]Ezan E.Pharmacokinetic studies of protein drugs: past, present and future[J].Adv Drug Deliv Rev, 2013, 65(8): 1065-1073.
[2]Zhang F, Liu M R, Wan H T.Discussion about several potential drawbacks of pegylated therapeutic proteins[J].Biol Pharm Bull, 2014, 37(3): 335-339.
[3]Kontermann R.Therapeutic proteins: strategies to modulate their plasma half-lives[M].Hoboken: John Wiley & Sons, 2012.
[4]Schlapschy M, Theobald I, Mack H,et al.Fusion of a recombinant antibody fragment with a homo-amino-acid polymer: effects on biophysical properties and prolonged plasma half-life[J].Protein Eng Des Sel, 2007, 20(6): 273-284.
[5]Schellenberger V, Wang C W, Geething N C,et al.A recombinant polypeptide extends thein vivohalf-life of peptides and proteins in a tunable manner[J].Nat Biotechnol, 2009, 27(12): 1186-1190.
[6]Yuen K C, Conway G S, Popovic V,et al.A long-acting human growth hormone with delayed clearance (VRS-317): results of a double-blind,
placebo-controlled, single ascending dose study in growth hormonedefcient adults[J].J Clin Endocrinol Metab, 2013, 98(6): 2595-2603.
[7]Zalevsky J, Chamberlain A K, Horton H M,et al.Enhanced antibody half-life improvesin vivoactivity[J].Nat Biotechnol, 2010, 28(2): 157-159.
[8]Andersen J T, Daba M B, Sandlie I.FcRn binding properties of an abnormal truncated analbuminemic albumin variant[J].Clin Biochem, 2010, 43(4): 367-372.
[9]Kenanova V E, Olafsen T, Salazar F B,et al.Tuning the serum persistence of human serum albumin domain III: diabody fusion proteins[J].Protein Eng Des Sel, 2010, 23(10): 789-798.
[10]Shpilman M, Niv-Spector L, Katz M,et al.Development and characterization of high affnity leptins and leptin antagonists[J].J Biol Chem, 2011, 286(6): 4429-4442.
[11]Levin A M, Bates D L, Ring A M,et al.Exploiting a natural conformational switch to engineer an interleukin-2 'superkine'[J].Nature, 2012, 484(7395): 529-533.
[12]徐瑞光, 贺进田, 贾锴, 等.Arg77和Glu80定点突变同时去除葡激酶中的T和B细胞抗原表位[J].微生物学报, 2011, 51(5): 692-703.
[13]Salvat R S, Choi Y, Bishop A,et al.Protein deimmunization via structure-based design enables efficient epitope deletion at high mutational loads[J].Biotechnol Bioeng, 2015, 112(7): 1306-1318.
[14]Grünewald J, Tsao M L, Perera R,et al.Immunochemical termination of self-tolerance[J].Proc Natl Acad Sci U S A, 2008, 105(32): 11276-11280.
[15]Grünewald J, Hunt G S, Dong L,et al.Mechanistic studies of the immunochemical termination of self-tolerance with unnatural amino acids[J].Proc Natl Acad Sci USA, 2009, 106(11): 4337-4342.
[16]Gauba V, Grünewald J, Gorney V,et al.Loss of CD4 T-cell-dependent tolerance to proteins with modifed amino acids[J].Proc Natl Acad Sci USA, 2011, 108(31): 12821-12826.
[17]Hardy L L, Wick D A, Webb J R.Conversion of tyrosine to the infammation-associated analog 3′-nitrotyrosine at either TCR-or MHC-contact positions can profoundly affect recognition of the MHC class I-restricted epitope of lymphocytic choriomeningitis virus glycoprotein 33 by CD8 T cells[J].J Immunol, 2008, 180(9): 5956-5962.
[18]Klechevsky E, Flamar A L, Cao Y,et al.Cross-priming CD8+T cells by targeting antigens to human dendritic cells through DCIR[J].Blood, 2010, 116(10): 1685-1697.
[19]Idoyaga J, Lubkin A, Fiorese C,et al.Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A[J].Proc Natl Acad Sci U S A, 2011, 108(6): 2384-2389.
[20]Gribenko A V, Patel M M, Liu J,et al.Rational stabilization of enzymes by computational redesign of surface charge-charge interactions[J].Proc Natl Acad Sci USA, 2009, 106(8): 2601-2606.
[21]Dombkowski A A.Disulfde by design: a computational method for the rational design of disulfde bonds in proteins[J].Bioinformatics, 2003, 19(14): 1852-1853.
[22]Mateo R, Luna E, Rincón V,et al.Engineering viable foot-and-mouth disease viruses with increased thermostability as a step in the development of improved vaccines[J].J Virol, 2008, 82(24): 12232-12240.

[专家介绍] 姚文兵:博士,教授,博士生导师。1983年毕业于南京药学院,2004年获中国药科大学微生物与生化药学博士学位。现任中国药科大学副校长,担任教育部高等学校药学类专业教学指导委员会主任委员,中国药师协会副会长、中国药学会生化与生物技术药物专业委员会委员,全国高等医学教育学会药学教育研究会副理事长兼秘书长,卫生部教材建设专家委员会委员,中华医学会医学教育分会常务理事;《药学教育》常务副主编、《药物生物技术》副主编,《中国药学年鉴》、《中国药科大学学报》等杂志编委。目前任国家食品药品监督管理局药品、保健食品评审委员,国家发改委药品价格决策专家委员会委员。科研工作主要研究领域为生物新药的研制、蛋白质类药物的分子改构和修饰研究,作为课题负责人主持承担国家自然科学基金重点项目、国家科技重大专项课题、国家863项目、国家自然科学基金面上项目等课题多项。完成了3个生物新药的临床前研究,其中1个获新药临床批件;申请专利22项,获授权发明专利14项,出版著作7部;近5年发表科研论文109篇,其中SCI论文65篇。要负责5项省部级抗体研究项目,授权抗体专利3项,公开抗体专利5项,发表SCI及中文核心论文40余篇。
Advances in Research on Design-based Optimization Strategy for the Drugability of Biological Macromolecules
TIAN Hong, YIN Jun, DAI Xin, NING Hongyu, CHEN Song, GAO Xiangdong, YAO Wenbing
(School of Life Science and Technology, China Pharmaceutical University, Nanjing 210009, China)
Biological medicines are in rapid development stage for now.However, some inherent characteristics of biological macromolecules restricts their drugability, so that a lot of biological macromolecules with therapeutic potential fail to be eventually developed into drugs.As a result, the development of biological medicines was seriously restricted.Bottleneck of biological medicine development has shifted from "the generation of new molecular" to "how to get a good physiological characteristics and the expected treatment effect of effective drug".In recent years, the research on reconstruction of biological macromolecular structures by rational design to optimize their drugability has developed rapidly.In this paper, the research progress on design-based optimizaion strategy for the drugability of biological macromolecules was reviewed.
biological macromolecule; drugability; rational design
R918
A
1001-5094(2015)04-0277-06
接受日期:2015-03-03
项目资助:国家自然科学基金(No.81430082);中央高校基本科研业务费重大研究计划引导项目(No.YD2014SK0002)
*通讯作者:姚文兵 教授;
研究方向:生物技术与生物制药;
Tel:025-83271218;E-mail:wbyao@cpu.edu.cn
