蒺藜苜蓿全基因组中WRKY转录因子的鉴定与分析
2014-11-17宋辉南志标
宋辉,南志标
兰州大学草地农业科技学院,兰州 730020
WRKY基因家族是一类只存在于植物的转录因子,主要参与植物体内转录调控和许多信号转导过程[1]。WRKY通常含有长约60个氨基酸序列的保守区域,该区域内一般含有保守的WRKYGQK七肽序列和紧随其后的保守锌指基序(Zinc finger motif)[2]。正是由于WRKYGQK保守七肽序列的存在,人们习惯上把含有这段保守序列的转录因子称为 WRKY转录因子或WRKY基因[3]。目前的研究发现,WRKY结构域的C末端主要存在两种不同类型的保守锌指基序[4],即C-X4-5-C-X22-23-H-X-H(C2H2)和C-X5-8-CX25-28-H-X1-2-C(C2HC)。根据WRKY结构域的数量和两种不同类型的保守锌指基序,一般将WRKY基因分为3种类型,即含有2个WRKY结构域的Ⅰ型基因、含有1个WRKY结构域和C2H2基序(Motif)的Ⅱ型基因和含有1个WRKY结构域和C2HC基序的Ⅲ型基因[2]。另外,WRKY转录因子通常与目标基因上游启动子区域的顺式作用元件W-box相结合起到转录调控的作用[2,3,5]。但是,有研究表明[6,7],WRKY转录因子还与SURE顺式作用元件结合发挥作用,因此推测SURE可能是另一类与WRKY转录因子相结合的重要顺式作用元件。
自 1994年 Ishiguro 和 Nakamura[8]从甘薯(Ipomoea batatasL.) 基因组中分离出WRKY基因的cDNA序列以来,研究者们相继从拟南芥(Arabidopsis thaliana)[2,9]、马铃薯(Solanum tuberosumL.)[10]、亚洲棉(Gossypium arboretumL.)[11]、水稻(Oryza sativaL.)[12]、大麦(Hordeum vulgareL.)[6]、小立碗藓(Physcomitrella patens)[13]、杨树(Populus tremula×Populus alba)[14]、向日葵(Helianthus annuusL.)[15]、黄瓜(Cucumis sativusL.)[16]和二穗短柄草(Brachypodium distachyonL.)[17]等植物基因组中鉴定出WRKY基因。在对这些WRKY基因进行功能验证的过程中,研究者们发现WRKY基因广泛参与各种生理生化反应,包括生物胁迫和非生物胁迫以及植物的发育[1,2]。例如,当植物受到伤害[18]、紫外线照射[19]、干旱、盐害、低温和高温[20,21]以及病原菌侵染[22]等胁迫时,WRKY基因的表达量都会上调。同时,WRKY基因家族在植物种子发芽[23]、植株衰老[24]、休眠[25]等方面也具有重要的生理应答。
既然WRKY基因家族参与如此多的调控,势必要求WRKY原始基因在漫长的进化过程中分化出不同功能的基因。基因重复(Gene duplication) 是植物在长期的进化过程中逐步形成的一种机制,主要是通过基因不等交换、逆转录转座或全基因组重复等途径产生一个与原基因相似的基因或碱基序列[26]。基因复制主要分为两种形式,一种是串联重复(Tandem duplication),一种是片段重复(Segmental duplication)。基因重复事件在基因扩张中具有重要的作用,尤其是串联重复,基因家族中一些不同功能的基因很有可能是由于串联重复产生的[27]。例如,对大豆(Glycine maxL.)[28]和毛果杨(Populus trichocarpa)[29]WRKY基因家族进行分析时发现,其数量众多的WRKY基因是通过串联重复产生的。
蒺藜苜蓿(Medicago truncatulaL.)作为优良的牧草在全世界范围内被广泛地种植。由于其具有基因组较小、二倍体、自花授粉、易于转化、再生时间较短等特点被作为豆科模式植物进行研究[30]。本研究利用生物信息学方法对蒺藜苜蓿全基因组中的WRKY基因家族的基因重复、染色体定位、系统发生、保守基序和 EST表达模式等进行分析,希望该研究结果能为以后在豆科植物的育种过程中提供重要的理论依据。
1 材料和方法
1.1 WRKY基因的鉴定
本研究以蒺藜苜蓿全基因组序列(Mt3.5v5) 为研究对象,基因组序列从蒺藜苜蓿公布测序结果的网站获得(http://www.medicago.org)。将下载到的蒺藜苜蓿蛋白质序列使用BLAST构建本地数据库,以Pfam数据库(http://pfam.janelia.org) 中WRKY结构域(PF03106)为查询序列进行BLAST搜索(P-value=10-3)[31,32]。利用 Clustal W 程序[33]对所确定的 WRKY序列进行多序列比对(Multiple sequence alignment),手工去除冗余序列和不完整读码框的序列。使用Pfam数据库对WRKY蛋白质序列进行结构域检测,去除不含WRKY结构域的序列。
1.2 WRKY基因的基因重复和染色体定位
利用本地 BLAST程序(P-value=10-10) 对蒺藜苜蓿WRKY基因家族进行基因重复分析[31,34]。基因重复事件必须同时满足以下条件[35]:(1) 在核苷酸序列比对区域中,相对短的核苷酸序列必须覆盖相对长的核苷酸序列的70%以上;(2) 在氨基酸序列比对区域中,两条氨基酸的相似度要达到70%以上。
蒺藜苜蓿WRKY基因在每条染色体上的定位信息由蒺藜苜蓿公布基因组序列网站中的 BLAST程序进行确认,其染色体物理定位图使用 MapInspect程序进行绘制。
1.3 WRKY基因家族的系统进化和基因结构分析
为了解蒺藜苜蓿WRKY基因的系统发生关系,通过Clustal X程序[36]对蒺藜苜蓿WRKY结构域进行多序列比对,使用 MEGA 4.0程序[37]构建系统进化树。系统进化树采用邻接法(NJ),Bootstrap值设置为1 000。
使用在线程序 PAL2NAL计算蒺藜苜蓿WRKYⅢ基因在进化中的选择压力。如果Ka/Ks>1,表示基因经受正选择压力;Ka/Ks=1,表示基因经受中性选择压力;Ka/Ks<1,表示基因受纯化选择压力。
先前的研究表明,WRKY结构域中主要含有两种不同类型的保守内含子[2]。本研究采用在线程序GSDS分析蒺藜苜蓿WRKY结构域的基因结构。
1.4 WRKY基序分析
在线程序 MEME[38]可以很好的分析基因的基序。本研究将蒺藜苜蓿 WRKY蛋白质序列提交到MEME网站预测WRKY基因的基序。在线程序MEME的参数设置为:(1) 基序重复的数量为“any”;(2) 基序的长度为6~200;(3) 预测基序的数量为20。
1.5 WRKY基因EST表达模式分析
为了预测蒺藜苜蓿WRKY基因的表达情况,本研究从NCBI网站的GenBank中下载了所有蒺藜苜蓿的EST序列。通过构建蒺藜苜蓿EST本地数据库,以蒺藜苜蓿WRKY基因为查询序列进行本地BLAST搜索(P-value=10-10)[34]。比对结果符合以下条件的被认为是假定的EST表达模式(EST expression profile):(1)在比对区域内,两序列的相似度在 96%以上;(2)在比对区域内,核苷酸序列的长度在200 bp以上。
2 结果与分析
2.1 WRKY基因的鉴定及分类
通过使用本地 BLAST程序对蒺藜苜蓿全基因组进行筛选,共分离到93条含有完整读码框的候选WRKY序列。经过Pfam数据库检测发现,这93条候选序列均含有 WRKY结构域。但是,其中 12个WRKY结构域中含不完整的WRKYGQK七肽序列或不完整的保守锌指基序。因此,本文把这些基因定义为非标准WRKY类型基因。与此相对,其他的WRKY基因(81个) 定义为标准WRKY类型基因。
依据对WRKY基因分类的标准[2],本研究发现,这93条候选序列中有20条序列含有2个WRKY结构域。其中,19个属于标准WRKY基因,定义为Ⅰ型,1个含有非标准的WRKY结构域。此外,其他73条候选序列均只含有一个WRKY结构域。其中,11个基因含有非标准 WRKY结构域,而另外 62个基因中有49个基因含有C2H2保守锌指基序,13个含有C2HC保守锌指基序,分别将其定义为Ⅱ型和Ⅲ型基因(表1,表 2)。
研究表明[2],Ⅱ型的WRKY基因可以细分为 5个亚组,即Ⅱa、Ⅱb、Ⅱc、Ⅱd和Ⅱe。本文以已公布的拟南芥WRKYⅡ型基因作为参考序列与蒺藜苜蓿WRKYⅡ型基因共同构建系统发育树,从而鉴别出蒺藜苜蓿WRKYⅡ型基因的5个亚组。由图1可知,蒺藜苜蓿WRKY基因(MtWRKY) 包括6个Ⅱa、8个Ⅱb、19个Ⅱc、8个Ⅱd和8个Ⅱe,其中Ⅱc被分为Ⅱc1(5个) 和Ⅱc2(14个) 两个分支。
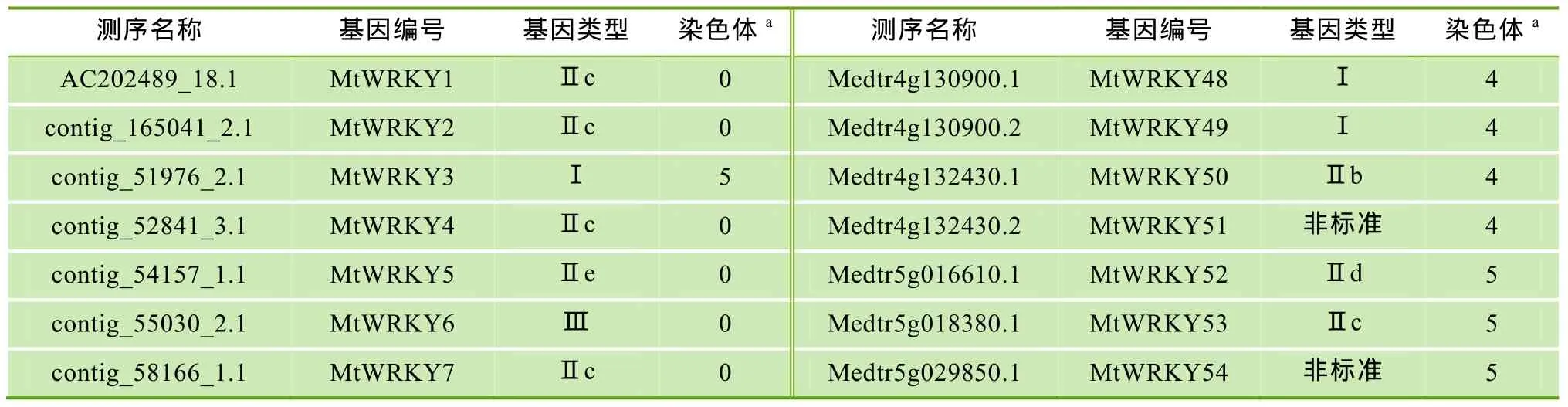
表1 蒺藜苜蓿WRKY基因的特征
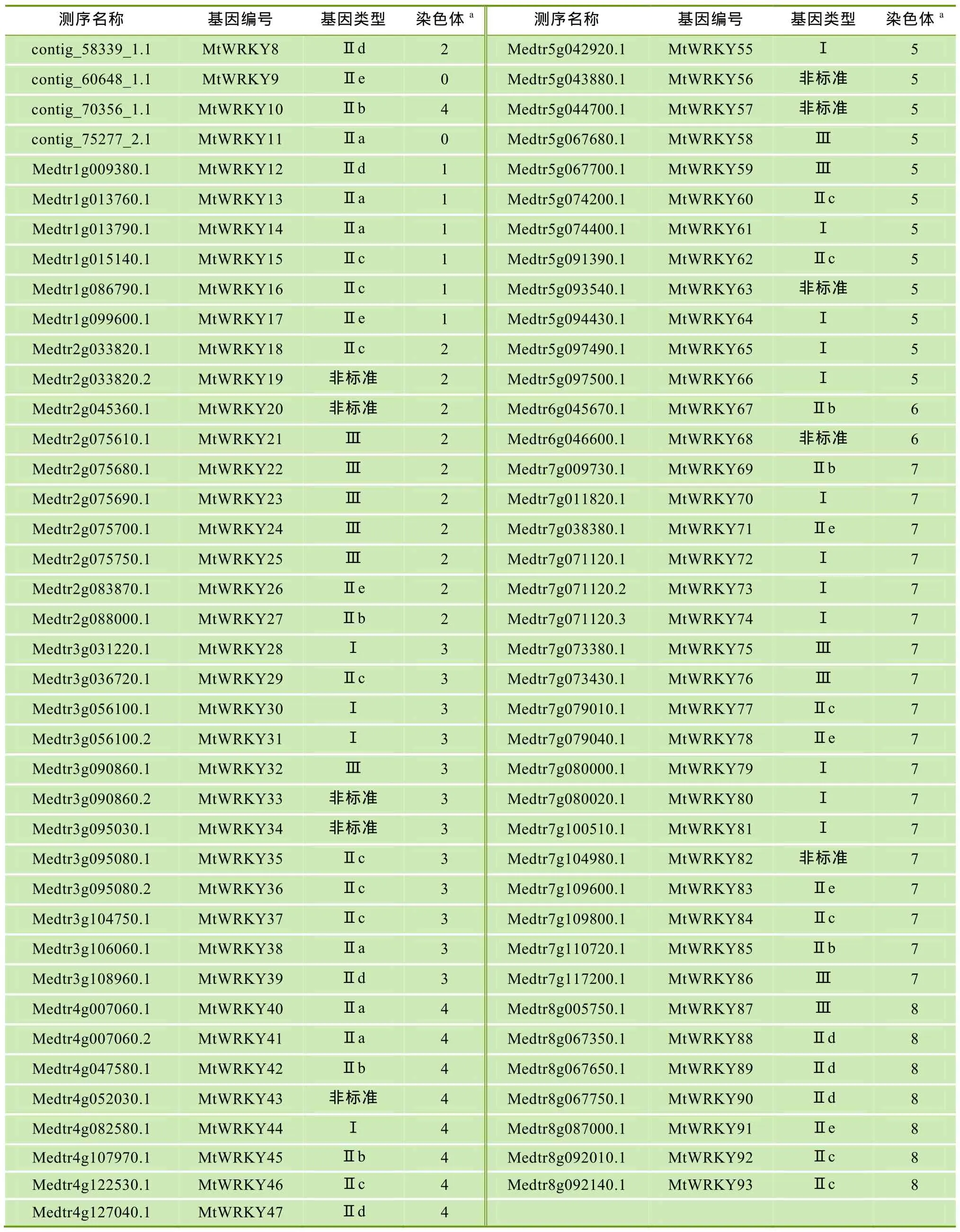
续表1
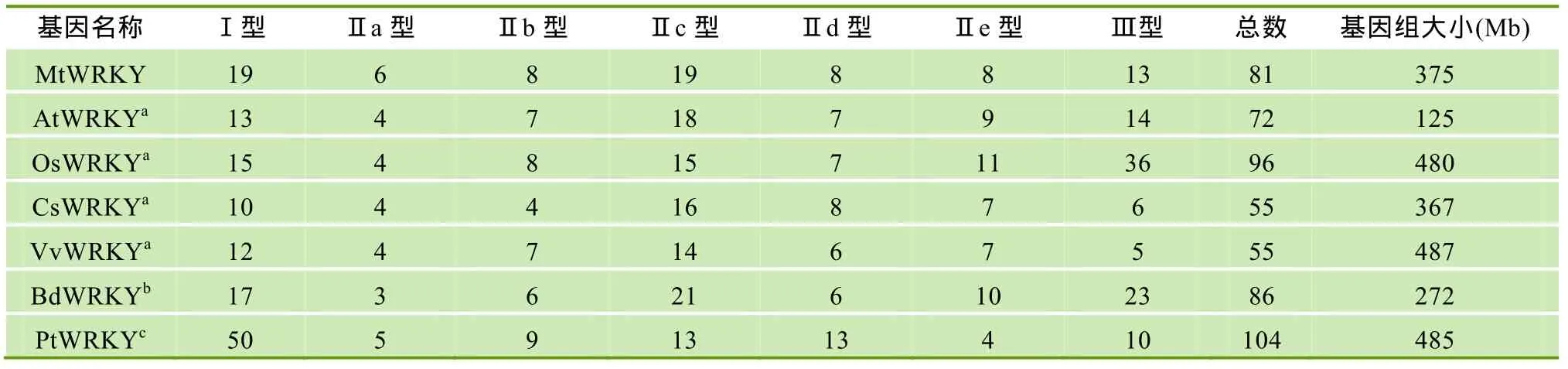
表2 7种植物中WRKY基因的分类和数目
2.2 WRKY的染色体定位和基因重复
蒺藜苜蓿93个WRKY基因在8条染色体上的物理定位信息见图2。由于蒺藜苜蓿现在尚未完成全基因组测序,所以有些基因暂时无法定位在具体的染色体上,本研究暂时将这些基因定位在假设的 0号染色体上。
由图2可知,蒺藜苜蓿WRKY基因在染色体上呈不均等分布。其中7号染色体上所含的WRKY基因最多,多达18个,而6号染色体上分布的WRKY基因最少,仅有2个。值得注意的是,分布在1号染色体上的6个WRKY基因均为Ⅱ型。另外,6个WRKYⅡ型基因和1个WRKYⅢ型基因分布在8号染色体上。有研究表明[4],Ⅲ型的WRKY基因的起源晚于Ⅱ型的WRKY基因。基于这种观点,结合蒺藜苜蓿WRKY基因在8号染色体上的物理定位,推测WRKYⅢ型基因可能是由于Ⅱ型基因经过基因突变而形成的。
根据Holub[39]对基因簇(Gene cluster) 的定义:在200 kb核苷酸序列区域内,含有3个或3个以上的基因定义为一个基因簇。最终,本研究在蒺藜苜蓿WRKY基因中发现了 6个基因簇共包括17个基因。其中,最大的一个基因簇含有5个基因分布在2号染色体上。此外,每个基因簇平均含有3.4个基因(表3)。
在蒺藜苜蓿WRKY基因中共检测到11个基因复制事件,涉及24个基因,占全部WRKY基因的26%。其中,4个基因属于片段重复,20个基因属于串联重复(图2,表3)。另外,单基因占全部基因的74%,表明在蒺藜苜蓿中这些WRKY基因的形成可能主要不是依赖基因重复完成的。
2.3 蒺藜苜蓿WRKY结构域多重比对分析
分别对蒺藜苜蓿3种类型的WRKY结构域进行多序列比对,结果显示,除了Ⅰc、Ⅱc和Ⅱd型的结构域在 WRKYGQK七肽序列结构上呈现多样性之外,其他类型的 WRKY结构域均含有一致序列WRKYGQK。其中,在Ⅰc型结构域中发现 Q突变成 R,形成 WRKYGRK七肽序列; 而在Ⅱd型结构域中Q突变成S,形成WRKYGSK七肽序列。除此之外,在Ⅱc型结构域中WRKYGQK七肽序列的一致性最差,该序列分别突变成以下 6种类型,即WKKYGEE、WRKYGEQ、WRKYGEK、WRKYGKK、WKKYEEK 和 WLKYDRK(图 3)。
众所周知,在WRKY结构域中除了含有保守的七肽序列WRKYGQK之外,在该七肽序列之后还包括一个保守的锌指基序(C2H2或者 C2HC)[2]。本研究结果表明,只有在Ⅲ型的 WRKY结构域中存在C2HC基序,在其他类型的 WRKY结构域中只含有C2H2基序。该结果与之前对水稻的研究结果略有不同,在水稻中发现个别In型的氨基酸序列中除了含有 C2H2之外还具有C2HC基序[12,40]。这或许与不同植物中该类型基因发挥不同的作用有关系。
2.4 蒺藜苜蓿WRKY基因系统发生关系分析
蒺藜苜蓿WRKY基因系统发育树显示(图 4a),虽然系统发育树中有些自举值(Bootstrap) 偏低,但是系统发育树根据 WRKY结构域的分类可以明显的分为 6个分支,这说明此系统发育树的拓扑结构基本正确。其中,Ⅰn和Ⅰc分支分别是Ⅰ型WRKY结构域两种类型的聚类(n表示多肽链N端的WRKY结构域,c表示多肽链C端的WRKY结构域),Ⅱ型结构域分别在Ⅱa、Ⅱb、Ⅱc、Ⅱd和Ⅱe分支上。此外,Ⅲ型的成员比较保守,全部分布在Ⅲ分支上(图4a)。
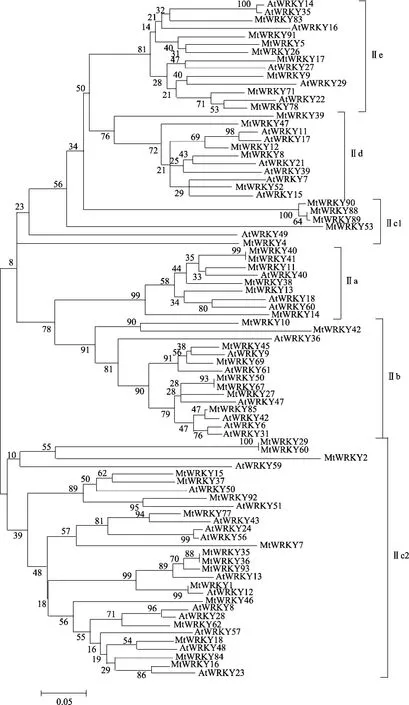
图1 拟南芥与蒺藜苜蓿WRKY结构域系统进化树
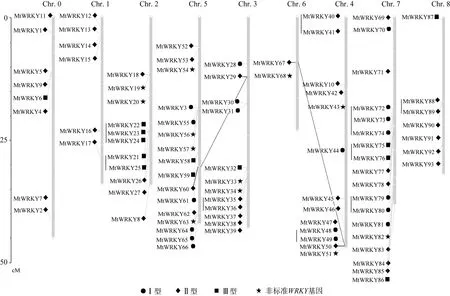
图2 蒺藜苜蓿WRKY基因在染色体上的定位信息

表3 蒺藜苜蓿基因组中WRKY基因的组成
Zhang等[4]通过研究 5种植物WRKY基因的系统发育关系发现,Ⅱa和Ⅱb聚集在一个大分支上,而Ⅱd和Ⅱe聚集在另一个大分支上。这个结果与本文分析蒺藜苜蓿 WRKY结构域构建的系统发育树相同。此外,本研究中Ⅱc与Ⅰc聚集在一个大分支上,甚至有一个Ⅱc型的基因(MtWRKY4) 聚集在Ⅰc分支上(图4a),这表明它们可能有共同的起源。但是,也有部分Ⅱc型的基因(MtWRKY2、29、53和60) 并没有聚集在Ⅱc分支上。其中,MtWRKY2、29和60聚集在Ⅰn分支上,而MtWRKY60聚集在Ⅱd和Ⅱe的大分支上(图4a),这表明Ⅱc型的基因可能存在不同的起源。
2.5 蒺藜苜蓿WRKY结构域基因结构分析

图3 蒺藜苜蓿WRKY结构域的氨基酸多序列比对
蒺藜苜蓿 WRKY结构域的基因结构主要分为两种类型:一种含有内含子,另一种不含内含子。由图4b可知,在系统进化树中聚集在Ⅰn的WRKY结构域均不含有内含子结构,这进一步支持了作为Ⅱc型的MtWRKY2、29 和60基因聚集在Ⅰn的结果。此外,虽然其他WRKY结构域含有不同长度的内含子,但是,根据内含子的类型可将其分为两大类。其中,Ⅱa和Ⅱb型的WRKY结构域具有同一个保守的内含子结构,而Ⅰc、Ⅱc、Ⅱe和Ⅲ拥有另外一种类型的保守内含子结构(图 4b)。但是,在Ⅱe中,MtWRKY9 却不含有内含子结构,这可能是由于其 WRKY结构域中的内含子在进化过程中丢失造成的。
2.6 蒺藜苜蓿WRKYⅢ类型基因选择压力分析
WRKY基因家族中Ⅲ型基因被认为是这3种类型的基因中最“年轻”的成员。为了检测哪种选择压力影响了蒺藜苜蓿WRKY基因家族中Ⅲ型基因的产生,本文对这些基因分别进行了同义替换率(Ka)和非同义替换率(Ks) 的计算。由图5可知,Ka/Ks<1,表示蒺藜苜蓿WRKY基因家族中Ⅲ型基因的进化经受纯化选择压力。由此可知,Ⅲ型的基因在结构上比较保守。
2.7 WRKY保守基序分析
通过对81个标准WRKY基序预测的结果显示,每一个类型的 WRKY所包含基序的类型各不相同(图6,表4)。通过使用pfam数据库检测这些基序的功能发现,只有motif1~6是WRKY结构域的组成部分,而其他的基序在数据库中暂无功能记录。进一步分析这些基序发现,在Ⅰn和Ⅰc型氨基酸序列中,含有WRKYGQK七肽序列的基序(motif1或motif2)两侧的基序的类型各不相同。这表明,虽然Ⅰ型氨基酸序列中存在两个WRKY结构域,但它们在起源或功能分化上可能完全不同。另外,虽然Ⅱa-e隶属于Ⅱ型基因的 5个亚型,但是每一个亚型的基序数量却不相同。例如,在Ⅱa和Ⅱb中均含有较多的基序,而Ⅱc和Ⅱe中却含有较少的基序(图6)。值得注意的是,在Ⅲ型氨基酸序列中存在一个有趣的现象,MtWRKY22、23、24和75这4个基因中的N末端和C末端分别存在一个相同的基序(motif20)。这种结构在其他类型的WRKY中尚未发现,这可能在功能域发挥作用时具有重要作用。
2.8 WRKY基因EST表达模式的分析
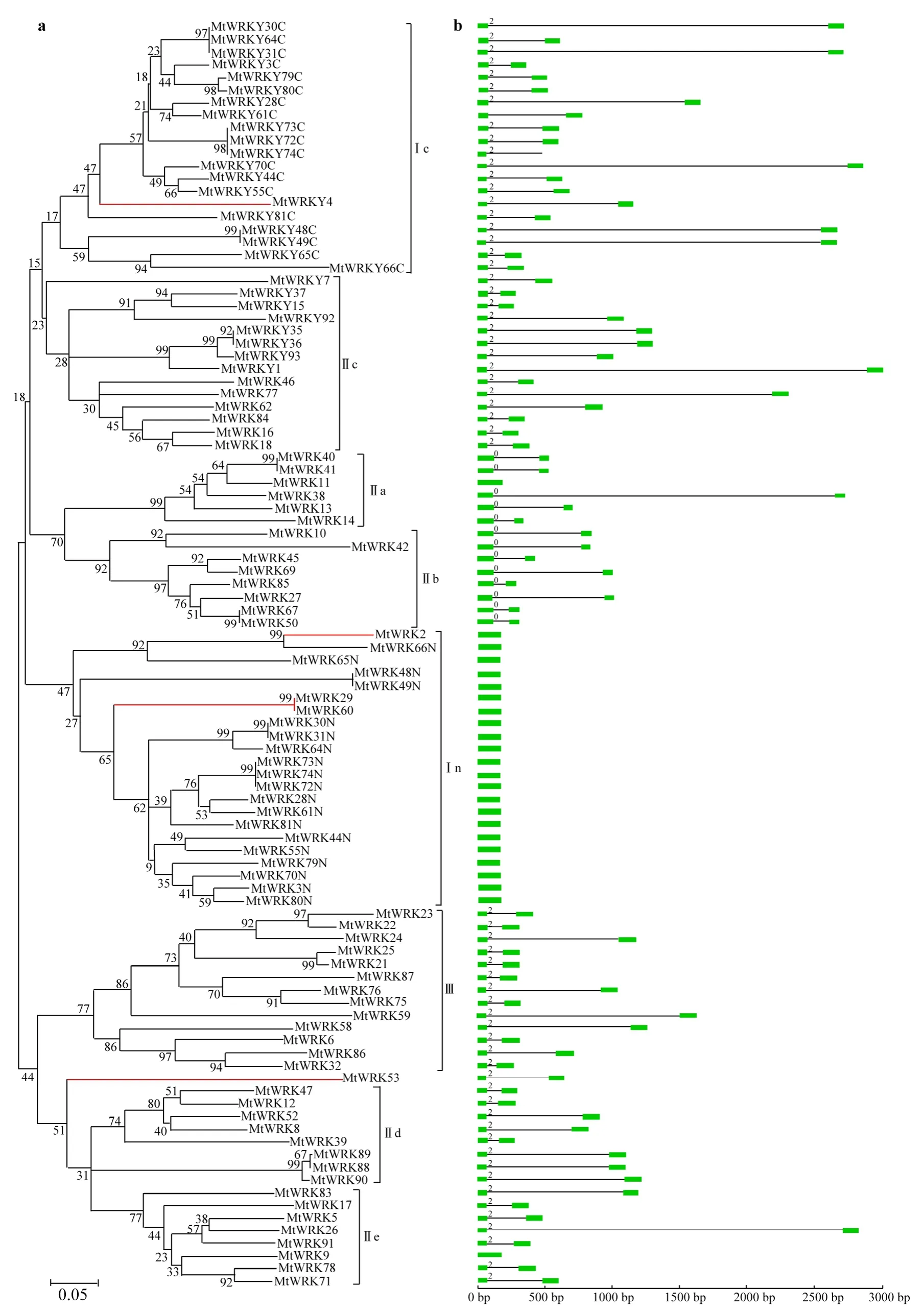
图4 蒺藜苜蓿中WRKY基因的系统发育树和基因结构
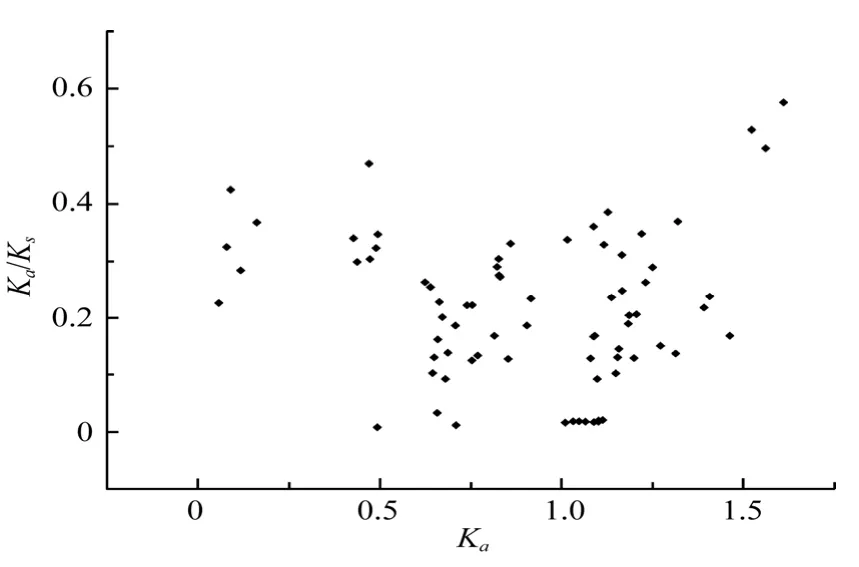
图5 WRKY基因家族中Ⅲ类型Ka/Ks与Ka的关系
表达序列标签(Expressing sequence tag,EST)被认为是一种研究基因表达模式最为有效的方法[41]。本文从GenBank中下载了全部250 000条蒺藜苜蓿的EST序列。通过使用蒺藜苜蓿93个WRKY基因与这些EST进行比对,结果发现,有60个WRKY基因在EST数据库的种子、芽、茎、腺毛、叶、根、组培细胞和组织或器官中找到了支持的表达模式(表5)。其中,WRKY基因与组织或器官中EST相匹配的表达模式最多,多达51个,而在种子的EST中只检测到3个相匹配的WRKY基因。值得注意的是,此处的组织或器官可能包括种子、芽、茎、腺毛、叶、根和组培细胞,但还可能包括未列出的组织或器官,如花等。此外,在这60个有表达模式的WRKY基因中,包含Ⅱ型的基因最多,高达 32个(53%),其次是15个( 25%)Ⅰ型的基因,再次是7个(12%)非标准的基因,而Ⅲ型的基因最少,只有 6个(10%)。这 3种类型的基因在 EST表达模式中所占的比例与这3种类型的基因在WRKY基因中所占的比例基本一致,这表明,在蒺藜苜蓿中,Ⅱ型的WRKY基因可能在植物体内生理生化反应过程中起主要作用。
3 讨 论
随着分子生物学尤其是测序技术的不断发展,越来越多的植物完成了全基因组序列的测定,如拟南芥[42]、水稻[43]和大豆[27]等。2011年,研究人员公布了作为主要豆科牧草的蒺藜苜蓿全基因组序列(Mt3.5v5) 的草图[30],这为人们从基因组水平上研究蒺藜苜蓿和进行分子育种提供了便利条件。

表4 蒺藜苜蓿中预测的WRKY基序的一致序列

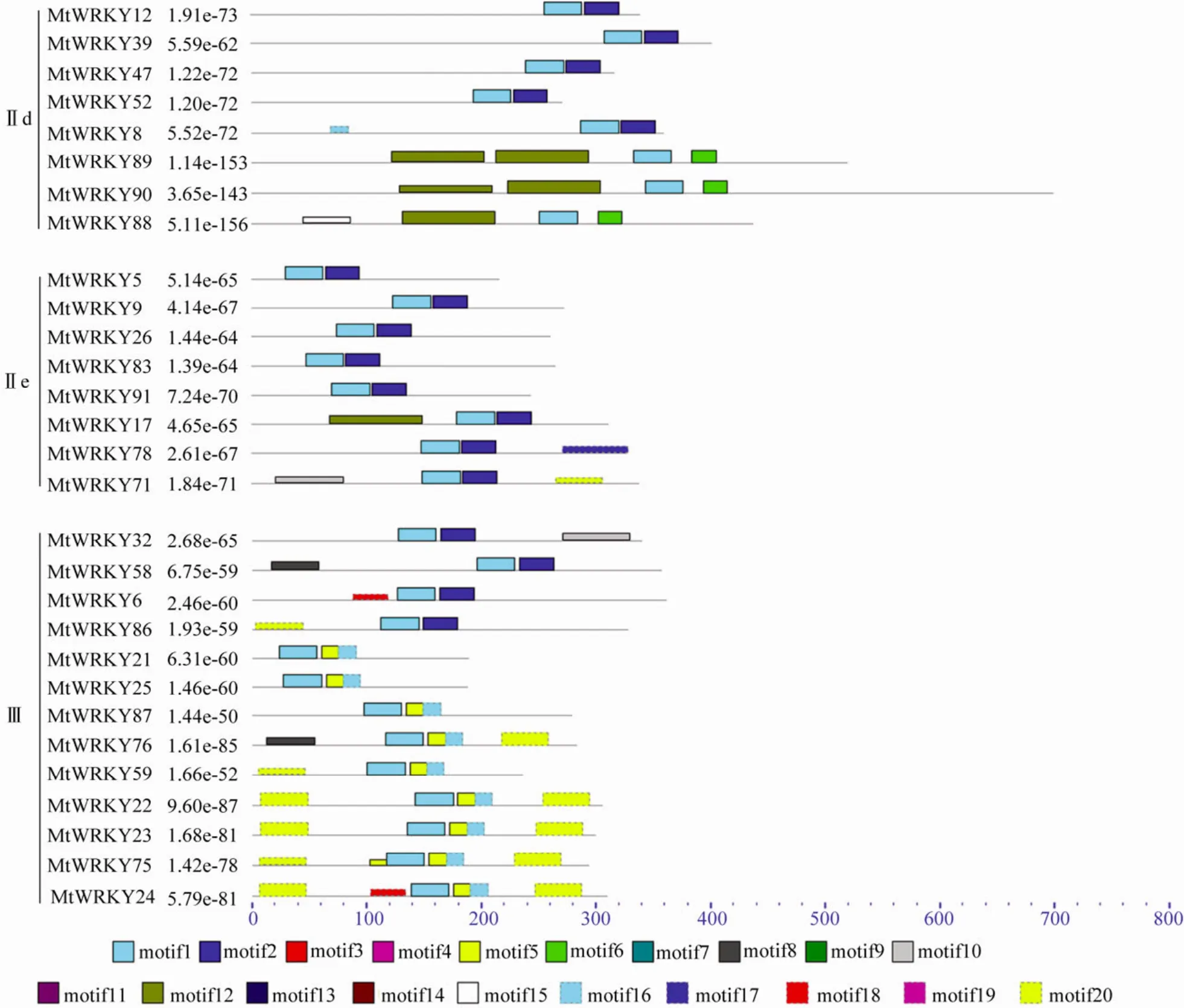
图6 蒺藜苜蓿WRKY中预测的基序
本研究最终从蒺藜苜蓿基因组中鉴定出 81个标准的WRKY基因。与其他植物相比较发现,植物基因组所含有WRKY基因的多少并不与其基因组的大小呈正比例关系。例如,葡萄(Vitis viniferaL.)的基因组大小为487 Mb,含有55个WRKY基因; 但是在蒺藜苜蓿 375 Mb的基因组中却分布有 81个WRKY基因; 而从二穗短柄草 272 Mb的基因组中共鉴定出86个WRKY基因(表2)。
基因重复事件是基因扩张的主要原因之一。在对水稻WRKY基因家族的研究中发现,80%的WRKY基因是由基因重复而来[44]。但是,本研究对蒺藜苜蓿WRKY基因家族的研究发现,只有24个WRKY基因发生了基因重复,仅占全部WRKY基因的26%,这个比例远低于水稻中发生基因重复的比率。我们推测,蒺藜苜蓿中WRKY基因家族出现这种现象的主要原因可能有以下3点:(1) 蒺藜苜蓿尚未完成全基因组序列的测定,因此,许多基因重复事件还无法进行检测;(2) 蒺藜苜蓿WRKY基因家族的成员可能不是通过基因重复事件进行扩张的。在对黄瓜的研究中已经发现,黄瓜WRKY基因家族中没有发现基因重复事件[16]。因此,在蒺藜苜蓿WRKY基因家族中可能仅存在低频率的基因重复事件;(3) 在面对各种生物和非生物胁迫过程时,蒺藜苜蓿WRKY基因家族为了降低能量的消耗,可能自行“消灭”体内冗余而没有功能的WRKY基因,基于这种保护体制可能使自身获得更有利的相对适合度。同样,在漫长的进化历史中,蒺藜苜蓿可以顺利的存活到现在也表明其体内数量有限的WRKY基因足够应付各种生物和非生物胁迫。
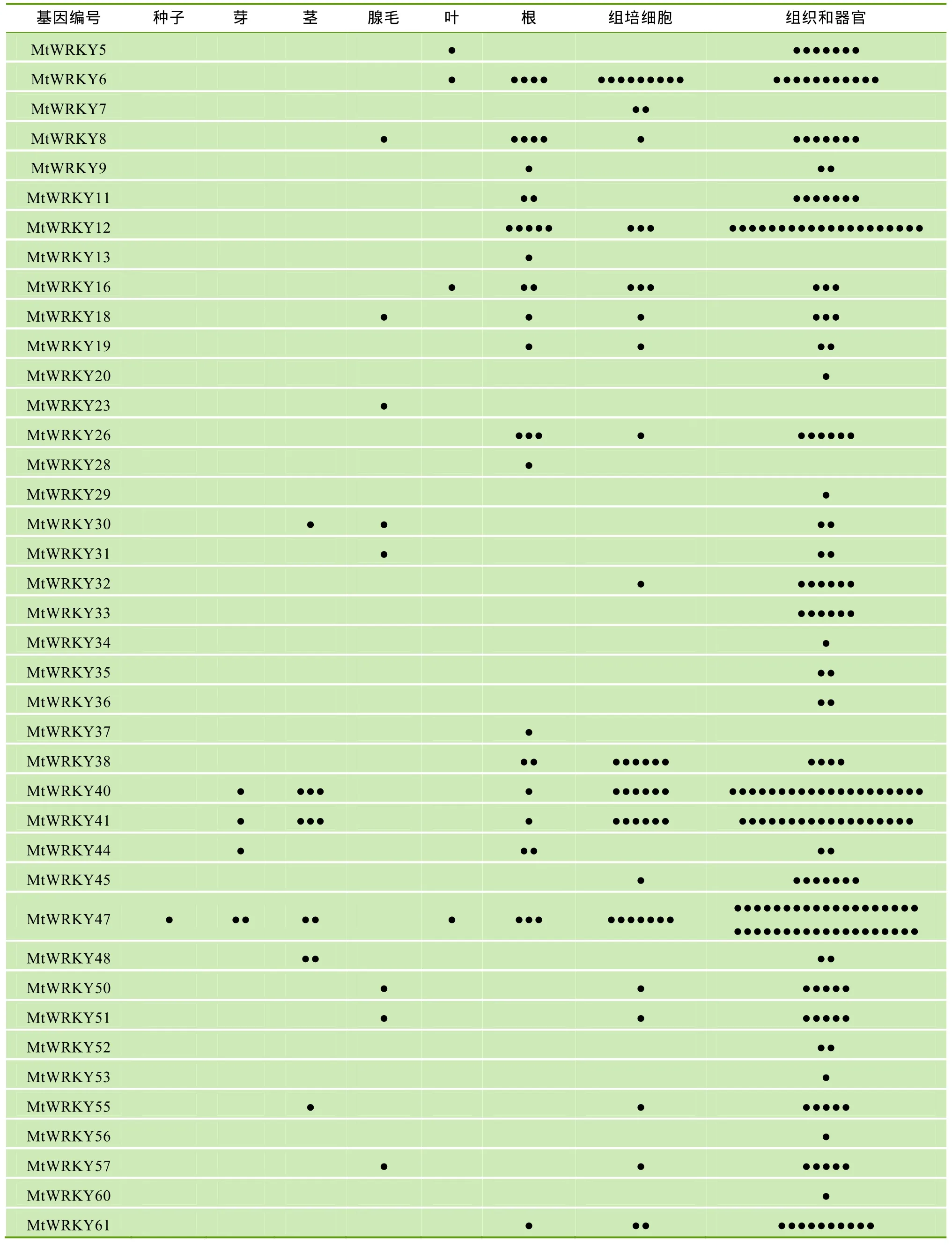
表5 WRKY基因的表达模式分析
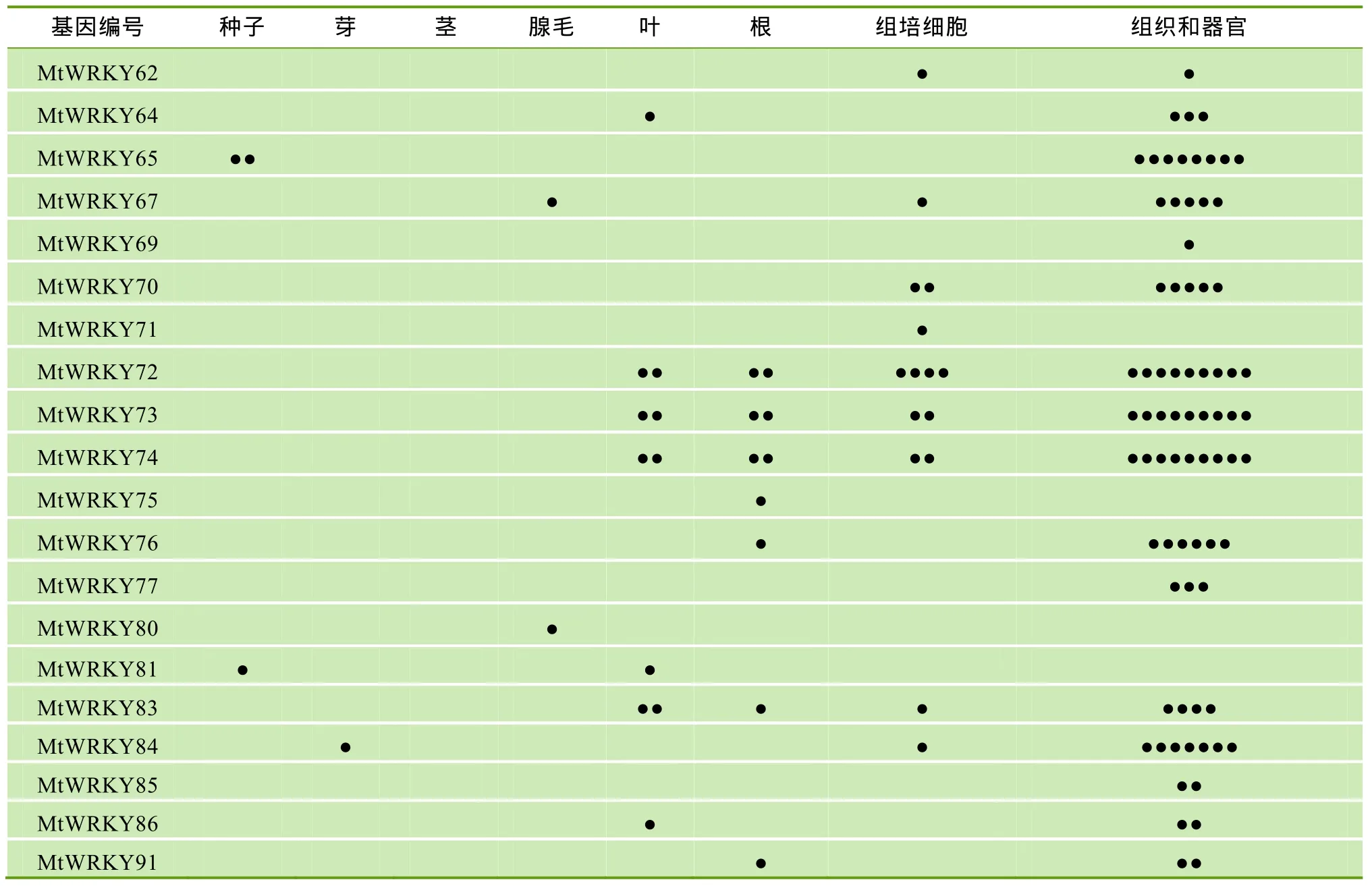
(续表5)
另外,蒺藜苜蓿WRKY基因在染色体上的物理定位的研究表明,WRKY基因在蒺藜苜蓿染色体上呈不均匀分布。但是,在对水稻[40]和毛果杨[29]的研究已经证实,WRKY基因主要分别集中在水稻的1号染色体和毛果杨的14号染色体上。这表明在水稻和毛果杨中这两条染色体上可能发生了高频率的基因重复事件。
通过分析蒺藜苜蓿 WRKY结构域的基因结构发现,蒺藜苜蓿WRKY结构域含有保守的内含子。2005年,Wu等[9]对水稻WRKY结构域研究发现,在Ⅰc、Ⅱc-e和Ⅲ型的结构域中,锌指结构第一个 C前面第5个氨基酸残基处(R) 是内含子的插入位点,并将这种内含子命名为R类型内含子,然而,在Ⅱa和Ⅱb型的结构域中,内含子的插入位点在锌指结构第2个C后的第6个氨基酸残基处(V),将其命名为V类型的内含子。但是,Ⅱa和Ⅱb型结构域中内含子的插入位点存在另外一种发现。在拟南芥和黄瓜的研究中发现[2,16],Ⅱa和Ⅱb型结构域内含子的插入位点位于锌指结构第2个C后的第4个氨基酸残基处(K)。本文对蒺藜苜蓿WRKY结构域的研究结果与在水稻中的研究结果一致(图 3)。对于这种保守内含子在 WRKY结构域不同位点的插入是否对WRKY基因的功能造成不同影响的研究尚未见报道。WRKY基因家族成员在调节植物体生理生化作用时各有分工,因此,保守内含子在WRKY结构域不同位点的插入是否影响WRKY基因的功能,可能是下一步研究的热点。
WRKY基因家族依据WRKY结构域的不同可以分为3种类型。目前研究者普遍认为[2,4],Ⅱ和Ⅲ型的WRKY结构域起源于Ⅰ型C端的结构域。Ⅱ型的C2H2保守锌指结构基序中的组氨酸(H) 被半胱氨酸(C) 替换后形成Ⅲ型中的C2HC保守锌指结构基序[40]。但是,本文在分析WRKY基序时发现,有些Ⅱ或Ⅲ型的结构域可能起源于Ⅰ型N端的结构域。例如,Ⅰ型N端3个基序(motif3,5和6) 出现在Ⅱ(MtWRKY2、29、60、88、89和 90) 和Ⅲ(MtWRKY 21、22、23、24、25、59、75、76 和 87) 型的基因中,而且更为重要的是在这些Ⅱ和Ⅲ型的基因中没有出现Ⅰ类型C端的结构域(图6,表4)。
更为有趣的是,在研究拟南芥WRKY结构域时发现,拟南芥Ⅲ型AtWRKY52/RRS1基因的WRKY结构域的 C末端含有 TIR-NBS-LRR基因(Toll/interleukin-receptor-nucleotide-binding site-leucine-rich repeat) 的结构域,而 TIR-NBS-LRR基因被证实参与植物对病原菌的免疫反应过程[45]。但是,本研究并没有发现类似的现象。为了尝试解释这种区别,本文一方面研究了蒺藜苜蓿WRKYⅢ型基因在进化过程中所受选择压力对基因进化的影响。通过计算Ka/Ks发现,蒺藜苜蓿Ⅲ型的WRKY基因在进化的过程中受纯化选择。这个结果与在黄瓜中的研究结果一致[16]。但是,在拟南芥中却发现,正选择压力在Ⅲ型基因的进化过程中起主导作用[16]。一般情况下,正选择压力更有益于基因的扩张或功能的分化,而纯化选择压力往往使基因变的更保守。因此,不难得出,在拟南芥中发生两种结构域的融合可能正是由于正选择压力的结果,相反,在蒺藜苜蓿或黄瓜中,“消极”的纯净化选择压力没有给该基因一个自我发展的机会,反而使该基因变的越来越保守。
另一方面,本文推测,相比与蒺藜苜蓿或黄瓜,拟南芥在生长周期中可能经历更多的病害。这势必要求在拟南芥中,抗病的基因在抵御病原菌的侵染过程中发挥更为重要的作用。由于病原菌的不断变异,同样会使抗病基因在结构上产生相应的变化,最终抗病基因与病原菌形成了协同进化,或许WRKYⅢ型的基因与 TIR-NBS-LRR基因的融合正是为了增强自身的抗病能力以适应不断变异的病原菌。
本研究利用EST序列分析了蒺藜苜蓿WRKY基因在生物和非生物胁迫下的表达模式,结果显示WRKY基因在蒺藜苜蓿体内具有广泛的表达,在组织或器官中的表达量最大,而在种子中的表达量最少,这可能与组织或器官经受比较多的胁迫而种子经受的胁迫比较少有关。但是,有报道证实,WRKY基因与种子发育有密切关系[22,46]。因此,本文推测,种子中的WRKY基因更倾向参与种子发芽的调控。
[1]Rushton PJ,Somssich IE,Ringler P,Shen QJ.WRKY transcription factors.Trends Plant Sci,2010,15(5):247– 258.
[2]Eulgem T,Rushton PJ,Robatzek S,Somssich IE.The WRKY superfamily of plant transcription factors.Trends Plant Sci,2000,5(5):199–206.
[3]Rushton PJ,Torres JT,Parniske M,Wernert P,Hahlbrock K,Somssich IE.Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes.EMBO J,1996,15(20):5690–5700.
[4]Zhang YJ,Wang LJ.The WRKY transcription factor superfamily:its origin in eukaryotes and expansion in plants.BMC Evol Biol,2005,5:1.
[5]Ciolkowski I,Wanke D,Birkenbihl RP,Somssich IE.Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function.Plant Mol Biol,2008,68(1-2):81–92.
[6]Mangelsen E,Kilian J,Berendzen KW,Kolukisaoglu UH,Harter K,Jansson C,Wanke D.Phylogenetic and comparative gene expression analysis of barley(Hordeum vulgare) WRKY transcription factor family reveals putatively retained functions between monocots and dicots.BMC Genomics,2008,9:194.
[7]Sun C,Palmqvist S,Olsson H,Borén M,Ahlandsberg S,Jansson C.A novel WRKY transcription factor,SUSIBA2,participates in sugar signaling in barley by binding to the sugar-responsive elements of theiso1promoter.Plant Cell,2003,15(9):2076–2092.
[8]Ishiguro S,Nakamura K.Characterization of a cDNA encoding a novel DNA-binding protein,SPF1,that recognizes SP8 sequences in the 5’ upstream regions of genes coding for sporamin and beta-amylase from sweet potato.Mol Gen Genet,1994,244(6):563–571.
[9]Wu KL,Guo ZJ,Wang HH,Li J.The WRKY family of transcription factors in rice andArabidopsisand their origins.DNA Res,2005,12(1):9–26.
[10]Dellagi A,Helibronn J,Avrova AO,Montesano M,Palva ET,Stewart HE,Toth IK,Cooke DEL,Lyon GD,Birch PRJ.A potato gene encoding a WRKY-like transcription factor is induced in interactins withErwinia carotovora subsp.atrosepticaandPhytophthora infestansand is coregulated with class I endochitinase expression.Mol Plant Microbe Interact,2000,13(10):1092–1101.
[11]Xu YH,Wang JW,Wang S,Wang JY,Chen XY.Characterization of GaWRKY1,a cotton transcription factor that regulates the sesquiterpene synthase gene(+)-deltacadinene synthase-A1.Plant Physiol,2004,135(1):507–515.
[12]Ross CA,Liu Y,Shen QJ.TheWRKYgene family in Rice(Oryza sativa).J Integr Plant Biol,2007,49(6):827–842.
[13]Rensing SA,Lang D,Zimmer AD,Terry A,Salamov A,Shapiro H,Nishiyama T,Perroud PF,Lindquist EA,Kamisugi Y,Tanahashi T,Sakakibara K,Fujita T,Oishi K,Shin IT,Kuroki Y,Toyoda A,Suzuki Y,Hashimoto S,Yamaguchi K,Sugano S,Kohara Y,Fujiyama A,Anterola A,Aoki S,Ashton N,Barbazuk WB,Barker E,Bennetzen JL,Blankenship R,Cho SH,Dutcher SK,Estelle M,Fawcett JA,Gundlach H,Hanada K,Heyl A,Hicks KA,Hughes J,Lohr M,Mayer K,Melkozernov A,Murata T,Nelson DR,Pils B,Prigge M,Reiss B,Renner T,Rombauts S,Rushton PJ,Sanderfoot A,Schween G,Shiu SH,Stueber K,Theodoulou FL,Tu H,Van de Peer Y,Verrier PJ,Waters E,Wood A,Yang L,Cove D,Cuming AC,Hasebe M,Lucas S,Mishler BD,Reski R,Grigoriev IV,Quatrano RS,Boore JL.ThePhyscomitrellagenome reveals evolutionary insights into the conquest of land by plants.Science,2008,319(5859):64–69.
[14]Levée V,Major I,Levasseur C,Tremblay L,MacKay J,Séguin A.Expression profiling and functional analysis ofPopulusWRKY23reveals a regulatory role in defense.New Phytol,2009,184(1):48–70.
[15]Guillaumie S,Mzid R,Mechin V,Léon C,Hichri I,Destrac-Irvine A,Trossat-Magnin C,Delrot S,Lauvergeat V.The grapevine transcription factor WRKY2 influences the lignin pathway and xylem development in tobacco.Plant Mol Biol2010,72(1–2):215–234.
[16]Ling J,Jiang WJ,Zhang Y,Yu HJ,Mao ZC,Gu XF,Huang SW,Xie BY.Genome-wide analysis of WRKY gene family inCucumis sativus.BMC Genomics,2011,12:471.
[17]Tripathi P,Rabara RC,Langum TJ,Boken AK,Rushton DL,Boomsma DD,Rinerson CI,Rabara J,Reese RN,Chen XF,Rohila JS,Rushton PJ.The WRKY transcription factor family inBrachypodium distachyon.BMC Genomics,2012,13:270.
[18]Hara K,Yagi T,Kusano T,Sano H.Rapid systemic accumulation of transcripts encoding a tobacco WRKY transcription factor upon wounding.Mol Gen Genet,2000,263(1):30–37.
[19]Izaguirre MM,Scopel AL,Baldwin IT,Ballaré CL.Convergent responses to stress.Solar ultraviolet-B radiation andManduca sextaherbivory elicit overlapping transcriptional responses in field-grown plants ofNicotiana longiflora.Plant Physiol,2003,132(4):1755–1767.
[20]Seki M,Narusaka M,Ishida J,Nanjo T,Fujita M,Oono Y,Kamiya A,Nakajima M,Enju A,Sakurai T,Satou M,Akiyama K,Taji T,Yamaguchi-Shinozaki K,Carninci P,Kawai J,Hayashizaki Y,Shinozaki K.Monitoring the expression profiles of 7000Arabidopsisgenes under drought,cold and high-salinity stresses using a full-length cDNA microarray.Plant J,2002,31(3):279–292.
[21]付乾堂,余迪求.拟南芥AtWRKY25、AtWRKY26和AtWRKY33在非生物胁迫条件下的表达分析.遗传,2010,32(8):848–856.
[22]Li J,Brader G,Palva ET.The WRKY70 transcription factor:a node of convergence for jasmonate-mediated and salicylate-mediated signals in plant defense.Plant Cell,2004,16(2):319–331.
[23]Johnson CS,Kolevski B,Smyth DR.TRANSPARENT TESTA GLABRA2,a trichome and seed coat development gene of Arabidopsis,encodes a WRKY transcription factor.Plant Cell,2002,14(6):1359–1375.
[24]Hinderhofer K,Zentgraf U.Identification of a transcription factor specifically expressed at the onset of leaf senescence.Planta,2001,213(3):469–473.
[25]Pnueli L,Hallak-Herr E,Rozenberg M,Cohen M,Goloubinoff P,Kaplan A,Mittler R.Molecular and biochemical mechanisms associated with dormancy and drought tolerance in the desert legumeRetama raetam.Plant J,2002,31(3):319–330.
[26]Nei M.Gene duplication and nucleotide substitution in evolution.Nature,1969,221(5175):40–42.
[27]Tan SL,Wu S.Genome wide analysis of nucleotide-binding site disease resistance genes inBrachypodium distachyon.Comp Funct Genomics,2012,2012:418208.
[28]Schmutz J,Cannon SB,Schlueter J,Ma J,Mitros T,Nelson W,Hyten D L,Song Q,Thelen JJ,Cheng J,Xu D,Hellsten U,May GD,Yu Y,Sakurai T,Umezawa T,Bhattacharyya MK,Sandhu D,Valliyodan B,Lindquist E,Peto M,Grant D,Shu S,Goodstein D,Barry K,Futrell-Griggs M,Abernathy B,Du J,Tian Z,Zhu L,Gill N,Joshi T,Libault M,Sethuraman A,Zhang XC,Shinozaki K,Nguyen HT,Wing RA,Cregan P,Specht J,Grimwood J,Rokhsar D,Stacey G,Shoemaker RC,Jackson SA.Genome sequence of the palaeopolyploid soybean.Nature,2010,463(7278):178–183.
[29]He HS,Dong Q,Shao YH,Jiang HJ,Zhu SW,Cheng BJ,Xiang Y.Genome-wide survey and characterization of theWRKYgene family inPopulus trichocarpa.Plant Cell Rep,2012,31(7):1199–1217.
[30]Young ND,Debelle F,Oldroyd GE,Geurts R,Cannon SB,Udvardi MK,Benedito VA,Mayer KF,Gouzy J,Schoof H,Van de Peer Y,Proost S,Cook DR,Meyers BC,Spannagl M,Cheung F,De Mita S,Krishnakumar V,Gundlach H,Zhou S,Mudge J,Bharti AK,Murray JD,Naoumkina MA,Rosen B,Silverstein KA,Tang H,Rombauts S,Zhao PX,Zhou P,Barbe V,Bardou P,Bechner M,Bellec A,Berger A,Berges H,Bidwell S,Bisseling T,Choisne N,Couloux A,Denny R,Deshpande S,Dai X,Doyle JJ,Dudez AM,Farmer AD,Fouteau S,Franken C,Gibelin C,Gish J,Goldstein S,Gonzalez AJ,Green PJ,Hallab A,Hartog M,Hua A,Humphray SJ,Jeong DH,Jing Y,Jocker A,Kenton SM,Kim DJ,Klee K,Lai H,Lang C,Lin S,Macmil SL,Magdelenat G,Matthews L,McCorrison J,Monaghan EL,Mun JH,Najar FZ,Nicholson C,Noirot C,O'Bleness M,Paule CR,Poulain J,Prion F,Qin B,Qu C,Retzel EF,Riddle C,Sallet E,Samain S,Samson N,Sanders I,Saurat O,Scarpelli C,Schiex T,Segurens B,Severin AJ,Sherrier DJ,Shi R,Sims S,Singer SR,Sinharoy S,Sterck L,Viollet A,Wang BB,Wang K,Wang M,Wang X,Warfsmann J,Weissenbach J,White DD,White JD,Wiley GB,Wincker P,Xing Y,Yang L,Yao Z,Ying F,Zhai J,Zhou L,Zuber A,Denarie J,Dixon RA,May GD,Schwartz DC,Rogers J,Quetier F,Town CD,Roe BA.TheMedicagogenome provides insight into the evolution of rhizobial symbioses.Nature,2011,480(7378):520–524.
[31]Altschul SF,Madden TL,Schäffer AA,Zhang J,Zhang Z,Miller W,Lipman DJ.Gapped BLAST and PSI-BLAST:a new generation of protein database search programs.Nucleic Acids Res,1997,25(17):3389–3402.
[32]Finn RD,Mistry J,Schuster-Böckler B,Griffiths-Jones S,Hollich V,Lassmann T,Moxon S,Marshall M,Khanna A,Durbin R,Eddy SR,Sonnhammer EL,Bateman A.Pfam:clans,web tools and services.Nucleic Acids Res,2006,34(1):D247–D251.
[33]Thompson JD,Higgings DG,Gibson TJ.CLUSTAL W:improving the sensitivity of progressive multiple sequence alignment through sequence weighting,position-specific gap penalties and weight matrix choice.Nucleic Acids Res,1994,22(22):4673–4680.
[34]Zhang Z,Schwartz S,Wagner L,Miller W.A greedy algorithm for aligning DNA sequences.J Comput Biol2000,7(1-2):203–214.
[35]Zhou T,Wang Y,Chen JQ,Araki H,Jing Z,Jiang K,Shen J,Tian D.Genome-wide identification of NBS genes injaponicarice reveals significant expansion of divergent non-TIR NBS-LRR genes.Mol Genet Genomics,2004,271(4):402–415.
[36]Thompson JD,Gibson TJ,Plewniak F,Jeanmougin F,Higgings DG.The CLUSTAL_X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools.Nucleic Acids Res,1997,25(24):4876–4882.
[37]Tamura K,Dudley J,Nei M,Kumar S.MEGA4:Molecular evolutionary genetics analysis(MEGA) software version 4.0.Mol Biol Evol,2007,24(8):1596–1599.
[38]Bailey TL,Elkan C.The value of prior knowledge in discovering motifs with MEME.Proc Int Conf Intell Syst Mol Biol,1995,3:21–29.
[39]Holub EB.The arms race is ancient history in Arabidopsis,the wildflower.Nat Rev Genet,2001,2(7):516–527.
[40]Xie Z,Zhang ZL,Zou X,Huang J,Ruas P,Thompson D,Shen QJ.Annotations and functional analyses of the riceWRKYgene superfamily reveal positive and negative regulators of abscisic acid signaling in aleurone cells.Plant Physiol,2005,137(1):176–189.
[41]Ohlrogge J,Benning C.Unraveling plant metabolism by EST analysis.Curr Opin Plant Biol,2000,3(3):224–228.
[42]Arabidopsis Genome Initiative.Analysis of the genome sequence of the flowering plantArabidopsis thaliana.Nature,2000,408(6814):796–815.
[43]Goff S A,Ricke D,Lan TH,Presting G,Wang R,Dunn M,Glazebrook J,Sessions A,Oeller P,Varma H,Hadley D,Hutchison D,Martin C,Katagiri F,Lange BM,Moughamer T,Xia Y,Budworth P,Zhong J,Miguel T,Paszkowski U,Zhang S,Colbert M,Sun WL,Chen L,Cooper B,Park S,Wood TC,Mao L,Quail P,Wing R,Dean R,Yu Y,Zharkikh A,Shen R,Sahasrabudhe S,Thomas A,Cannings R,Gutin A,Pruss D,Reid J,Tavtigian S,Mitchell J,Eldredge G,Scholl T,Miller RM,Bhatnagar S,Adey N,Rubano T,Tusneem N,Robinson R,Feldhaus J,Macalma T,Oliphant A,Briggs S.A draft sequence of the rice genome(Oryza sativaL.ssp.japonica).Science,2002,296(5565):92–100.
[44]Ramamoorthy R,Jiang SY,Kumar N,Venkatesh PN,Ramachandran S.A comprehensive transcriptional profiling of the WRKY gene family in rice under various abiotic and phytohormone treatments.Plant Cell Physiol,2008,49(6):865–879.
[45]Deslandes L,Olivier J,Peeters N,Feng DX,Khounlotham M,Boucher C,Somssich I,Genin S,Marco Y.Physical interaction between RRS1-R,a protein conferring resistance to bacterial wilt,and PopP2,a type Ⅲ effector targeted to the plant nucleus.Proc Natl Acad Sci USA,2003,100(13):8024–8029.
[46]Wenck AR,Conger BV,Trigiano RN,Sams CE.Inhibition of somatic embryogenesis in orchardgrass by endogenous cytokinins.Plant Physiol,1988,88(4):990–992.
