CuI催化溴代苯与L-缬氨酸的反应机理的研究
2014-08-07潘晓晓邹良明岳文喜李来才
潘晓晓, 邹良明, 岳文喜, 李来才∗
(1.四川师范大学化学与材料科学学院,四川成都610066; 2.四川工商职业技术学院,四川都江堰611837)
有机含氮化合物是一类非常重要的化合物,C—N键不仅在许多天然的富有生理活性的物质中存在,在现代社会工业化生产出的合成物中也常常出现.作为不可或缺的重要组分在一些结构简单的含氮化合物基础上构建新的C—N键,对合成含有这种结构单元的天然产物、药物、农药、材料等非常重要,因此这一直是有机化学家重点研究的课题之一[1].C—N交叉偶联起先是以Ni、Pd为催化剂[2],近年来人们发现Cu有很好的催化性能,并且具有更廉价更环保等优势,铜催化的C—N交叉偶联反应已成为有机合成中构建C—N键的主要途径[3-4],如今经过科学家们的不断改进,铜催化的C—N交叉偶联反应所需的条件已变得更加温和、简单,使其应用范围进一步扩大[5].然而,在这一领域中仍然存在着一些有待于更进一步研究的课题.近年来对偶联反应机理的理论研究工作开展得比较活跃[6-7],H.Z.Yu等[8]研究了亚铜催化的碳杂偶联反应机理,我们课题组研究过铜催化下N-(邻氯苯基)苯甲酰胺分子内O-芳基化偶联反应[9].它在对催化剂的优化、反应机理的描述和发现新的反应通道方面有着很大的优势,理论研究偶联反应微观机理能够更好地了解催化剂作用的本质,解释实验现象.
2009年Masanari团队的F.Ullmann[10]研究发现,用CuI催化剂能够催化溴代苯和L-缬氨酸发生C—N偶联反应.本文对这一典型的C—N偶联反应的微观反应特征进行了详细的机理研究,以了解这一合成反应的微观历程,为相关的实验及新问题的解决提供一些参考.用CuI催化剂能够催化溴代苯和L-缬氨酸发生C—N偶联反应,脱去HX,形成N-芳基-α-氨基酸,反应式如图1所示.
1 计算方法
采用密度泛函理论(DFT)[11-12],在B3LYP/6-311+G(d)基组水平上(Cu采用赝势基组LanL2DZ,卤素Cl、I采用赝势机组MIDIX)对CuX催化L-缬氨酸(N-芳基-α-氨基酸)与溴苯反应生成N-芳基-α-氨基酸过程中所有反应物、中间体、过渡态和产物进行了优化,并在相同基组水平下进行了频率计算,各过渡态都有唯一虚频,通过振动分析进一步确认了过渡态的真实性.通过反应中各物质相对能量比较,分析了该反应的速率决定步骤以及主要反应通道.同时在此基组水平上运用自然键轨道[13](NBO)分析方法分析了分子轨道间相互作用;用AIM 2000程序包[14]计算了相应的成键临界点(BCP)和成环临界点(RCP)电荷密度,分析了成键特征,所有计算均采用Gaussian 09程序[15].

图1 CuI催化的L-缬氨酸与溴代苯C—N偶联反应Fig.1 Copper-catalyzed C—N coupling reaction of aryl halides with α-amino acids
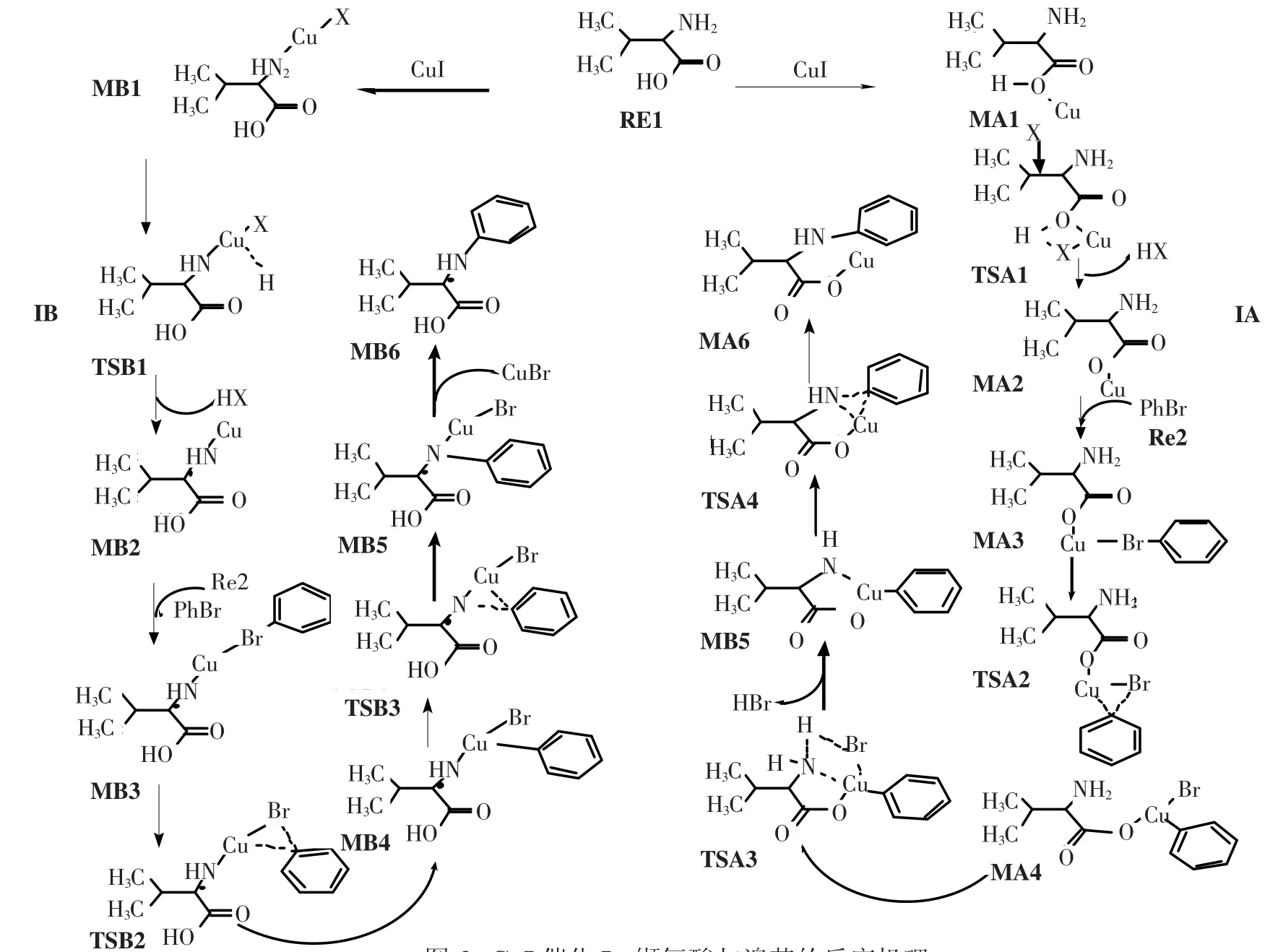
图2 CuI催化L-缬氨酸与溴苯的反应机理Fig.2 Detailed route of CuI-catalyzed reaction between aryl halides and α-amino acids
2 计算结果与讨论
图2所示为CuI催化L-缬氨酸(α-氨基酸)与溴苯反应生成N-芳基-α-氨基酸的反应机理.由反应物RE1与CuI结合开始得到了2条不同的反应通道,其中路径IA是CuI连到了α-氨基酸的羧基氧上开始的;而路径IB则是CuI连到了α-氨基酸的氨基上开始的.下面分别介绍这2条路径的反应机理.
2.1 路径IA反应机理分析表1为路径IA中各化合物的能量及校正后各物质的相关能量.该反应机理中所有反应物、中间体、过渡态和产物的优化分子构型以及部分键临界点(BCP)电荷密度(图中圆括号内)如图3所示.

表1 反应路径IA中各驻点能量E、相对能量Erel和过渡态的振动频率vTable 1 Energies E,relative energies Erel,and frequencies v of the compounds in the reactions for IA
在路径IA中,反应的第一步是催化剂CuI中的Cu原子与反应物α-氨基酸中的O(7)活化络合,得到中间体MA1.MA1中Cu—I键从反应物的键长0.230 1 nm增长到0.237 2 nm,电荷密度从0.092 3 a.u.减少至0.071 4 a.u.,Cu—O键长为0.198 9 nm,电荷密度为0.084 1 a.u.,H—O(1)键几乎没有变化.表明Cu与O有成键,RE1与CuI生成MA1.NBO计算分析得知,在中间体MA1中,BD(1)O(7)—H(17)→LP∗(8)Cu、BD(1)C(4)—O(7)→LP∗(8)Cu间的二阶稳定化能分别为35.55和20.31 kJ/mol,表明它们之间存在轨道间相互作用.从表1能量数据可以看出,中间体MA1的能量比α-氨基酸与催化剂CuI能量之和低82.89 kJ/mol,表明中间体MA1易形成并稳定存在.MA1到TSA1过程中,H原子从O(1)上往I上迁移.H—I键长为0.166 0 nm,电荷密度为0.134 3 a.u.;Cu—I键略有减弱,键长增长为0.247 9 nm.Cu—O(1)键长为0.190 5 nm,电荷密度为0.098 6 a.u.,此键略有减弱.MA2中Cu—O(1)键长为0.208 2 nm,电荷密度为0.065 1 a.u.;成键略有减弱.MA1→TSA1→MA2过程中,CuI连接到O(1)上,H从O上经过过渡态到Cu上,进而和I结合并脱去HI.RE2中Br—C(1)键长为 0.191 9 nm,电荷密度为0.158 5 a.u..生成中间体MA3后,Cu—O(1)键长为0.191 0 nm,电荷密度为0.098 1 a.u.,略有增强,Br—C(1)键长为0.196 0 nm,电荷密度为0.145 2 a.u.,此键略有减弱,Br—Cu键长为0.233 5 nm,电荷密度为0.069 0 a.u.,说明已经形成了中间体MA3.MA3到TSA2过程中,C(1)—Cu键有一定的成键,键长为0.196 9 nm,电荷密度为0.103 1 a.u.,C(1)—Br键长为0.261 7 nm,此一步表现了苯环从 Br上往 Cu上过渡的状态.在TSA2到MA4过程中,C(1)—Cu键长0.196 9 nm减小到0.193 6 nm,电荷密度增加到0.117 1 a.u.,此键进一步增强成键,同时Cu—N也有一定的成键,键长为0.203 8 nm,电荷密度为0.082 0 a.u.,形成MA4.在MA3→TSA2→MA4过程中,苯环从Br上过渡到 Cu上,同时 Cu—N成键.在 MA4到TSA3过程中,Br已经从Cu上往H(1)上迁移.H(1)—Br成键键长为0.148 8 nm,N—H(1)键减弱到键长为0.181 2 nm,电荷密度为0.044 2 a.u.,同时N—Cu键长为0.190 9 nm,电荷密度0.114 4 a.u..在TSA3到MA5过程中,HBr的进一步离去,O—Cu键长从0.184 6 nm增长到0.186 1 nm,电荷密度从0.115 8 a.u.减小到0.111 9 a.u.,此键有所减弱,Cu—C键长从0.191 6 nm减小到0.188 0 nm,电荷密度从0.123 2 a.u.增加到0.134 4 a.u.,此键略有加强,Cu—N键长从0.190 9 nm减小到0.186 8 nm,电荷密度从0.114 9 a.u.增加到0.124 7 a.u.,此键有所加强;进一步验证了HBr的离去,利于Cu—C(1)和Cu—N的成键.在MA4→TSA3→MA5过程中,Cu—N键进一步增强,HBr离去.在 MA5到 TSA4过程中,C(1)—Cu键长从0.188 0 nm增长到0.190 4 nm,电荷密度从0.134 4 a.u.减小到0.116 0 a.u.,此键减弱.N—C(1)在中间体 MA5中未成键,到 TSA4有成键,键长为0.205 4 nm,电荷密度为0.081 1 a.u..表明了苯环在向N上靠近,有一定的成键.在TSA4到MA6过程中,N—C(1)键长0.205 4 nm减少到0.145 7 nm,电荷密度从0.081 1 a.u.增加到0.301 4 a.u.,说明N与苯环已经成键.O—Cu键长从0.190 3 nm增加到0.212 1 nm,电荷密度从0.100 0 a.u.减小到0.066 4 a.u.,表面 Cu原子有脱离的趋势,在MA5→TSA4→MA6过程中,苯环从Cu原子转移到N原子上,并且Cu—N键断开.
2.2 路径IB反应机理分析表2为路径IB上各化合物能量以及校正后各物质的相关能量.该反应机理中所有反应物、中间体、过渡态和产物的优化分子构型以及部分键临界点(BCP)电荷密度(图中圆括号内)如图4所示.
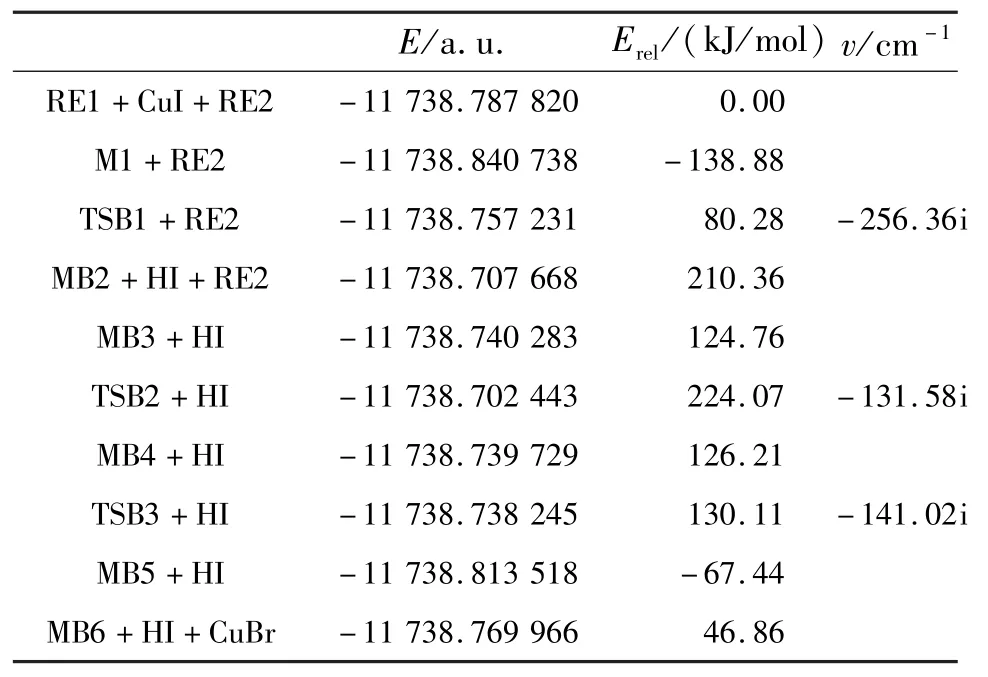
表2 反应路径IB中各驻点能量E、相对能量Erel和过渡态的振动频率vTable 2 Energies E,relative energies Erel,and frequencies v of the compounds in the reactions for IB
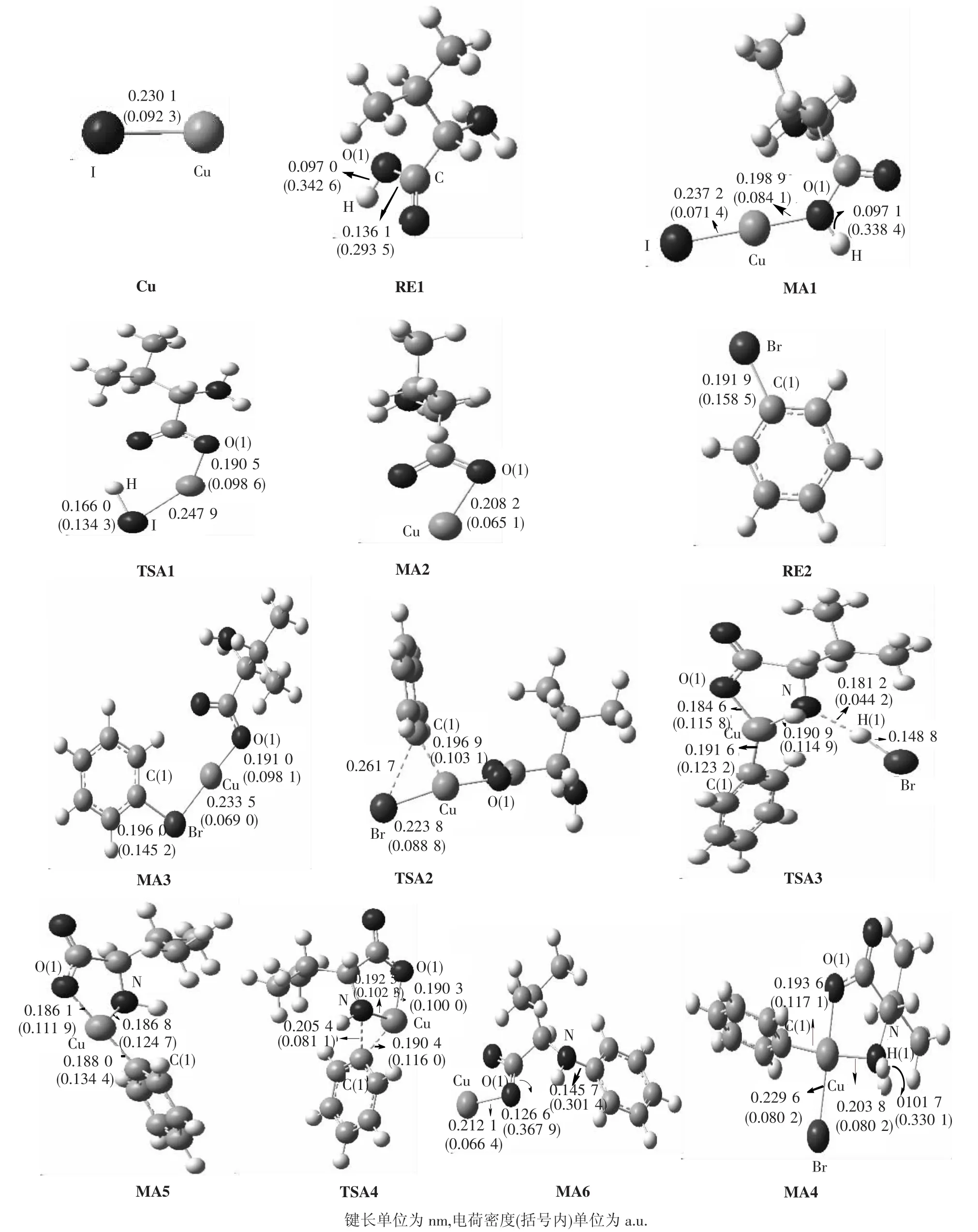
图3 路径IA上化合物的构型参数Fig.3 Geometric parameters of the compounds in pathway IA

图4 路径IB上化合物的构型参数Fig.4 Geometric parameters of the compounds in pathway IB
在路径IB中,反应的第一步是催化剂CuI中的Cu原子与反应物α-氨基酸的中的N(8)活化络合,得到中间体 MB1.CuI中 Cu与 I键长为0.230 1 nm,电荷密度为0.092 3 a.u.,形成中间体MB1时Cu与I键长增长到0.238 5 nm,电荷密度减少到0.089 8 a.u.,同时Cu与N键长为0.198 3 nm,表面Cu与N之间有成键.NBO计算分析得知,在中间体MB1中,BD(1)N(8)—H(18)→LP∗(6)Cu、BD(1)N(8)—H(19)→LP∗(6)Cu间的二阶稳定化能分别为35.59和35.51 kJ/mol,表明它们之间存在轨道间相互作用.从表2能量数据可以看出,中间体MB1的能量比α-氨基酸与催化剂CuI能量之和低138.88 kJ/mol,表明中间体MB1易形成并稳定存在,而且为后续反应提供潜热.中间体MB1上的H(1)原子经过渡态TSB1迁移到I原子上并且脱去形成中间体MB2.在MB1到TSB1过程中,Cu—N键长从0.198 3 nm减小到0.181 1 nm,电荷密度达到了0.140 8 a.u.,说明了Cu与N进一步成键,同时N上H(1)已经转移到Cu上,Cu—N键长为0.145 1 nm,电荷密度为0.131 7 a.u.,表面了Cu—N配位键的形成.另外Cu—Br键长从0.238 5 nm增长到0.240 2 nm,电荷密度从0.089 8 a.u.减小到0.081 4 a.u.,表明Cu—Br键在进一步减弱,随着HI的离去,中间体MB2甲基上的H和Cu有轻微的成键,其电荷密度为0.997 7 a.u.,伴随着Cu—N键的减弱,键长增长到0.184 1 nm.RE2的Br原子与MB2的Cu原子络合成键形成MB3.形成MB3后Br—C(1)键长伸长到0.195 1 nm,电荷密度为0.148 7 a.u.,同时 Br—Cu键长为0.234 4 nm,电荷密度为0.066 4 a.u.,Cu—N键略有增强,键长为0.138 9 nm,电荷密度为0.129 9 a.u..在中间体MB3上的苯环经过渡态TSB2从I原子上迁移到Cu原子上形成中间体MB4,在过渡态TSB2中Br—C(1)键增大到0.218 8 nm,电荷密度减少为0.090 7 a.u.,表明Br—C(1)键有所减弱,Br—Cu键长从0.234 4 nm到0.233 5 nm,电荷密度从0.064 4 a.u.到0.069 8 a.u.,此键几乎不变,Cu原子与C(1)有一定的成键,键长为0.206 7 nm,电荷密度为0.083 1 a.u.,表明了苯环有从Br原子上往Cu原子上的迁移.在中间体MB4中Cu—C(1)键进一步加强,表明Cu—C(1)进一步成键,苯环已经连接到了Cu原子上.中间体MB4上的苯环经过渡态TSB3从Cu原子上迁移到N原子上形成中间体MB5,随后CuBr脱去形成产物.

图5 反应过程各驻点能级变化图Fig.5 The diagram of relative energies along the channels of the reactions
2.3 反应过程能级变化分析反应过程各驻点能级变化如图5所示.从图5中可以看出,路径IA和路径IB经过L-缬氨酸(N-芳基-α-氨基酸)和催化剂CuI活化络合成稳定的中间体MA1和MB1,该过程中体系分别放出热量82.9和138.9 kJ/mol,为后续反应提供一定的潜热.路径IA中催化反应速率控制步骤是MA4→TSA3→MA5过程,反应活化能为282.2 kJ/mol;路径IB中催化速率控制步骤是MB1→TSB1→MB2过程,反应活化能为219.2 kJ/mol,由以上比较结果可以看出,IB通道具有较低的活化能,即IB通道为整个反应的最优反应通道,通道路径为RE1→MB1→TSB1→MB2→MB3→TSB2→MB4→TSB3→MB5→MB6.
3 结论
采用密度泛函理论在B3LYP/6-311+G∗基组水平上对CuI催化L-缬氨酸(α-氨基酸)与溴苯反应生成N-芳基-α-氨基酸的反应机理进行了理论研究,研究发现了2条可行的反应通道,路径IA:RE1→MA1→TSA1→MA2→MA3→TSA2→MA4→TSA3→MA5→TSA4→MA6;速率控制步骤为MA4→TSA3→MA5过程,反应活化能为282.2 kJ/mol;路径 IB:RE1→MB1→TSB1→MB2→MB3→TSB2→MB4→TSB3→MB5→MB6,速率控制步骤为MB1→TSB1→MB2过程,活化能为219.2 kJ/mol.通过比较各反应通道中控制步骤的活化能,得出IB通道具有较低的活化能,即IB通道为整个反应的最优反应通道,该通道的第一步活化络合过程能量降低了138.9 kJ/mol,为其速率控制步骤MB1→TSB1→MB2提供了一定的潜热,使反应易于进行,生成N-芳基-α-氨基酸.同时还计算了用CuCl催化的路径IB的控制步骤活化能为229.4 kJ/mol,高于CuI催化的活化能(219.2 kJ/mol),反应速率降低,这与文献报道是一致的.
[1]王晔峰,曾京辉,崔晓瑞.铜催化C—N交叉偶联反应的研究进展[J].有机化学,2010,30(2):181-199.
[2]Murahashi S I,Davies S G.Transition metal catalyzed reactions[J].Blackwell Science,1999,5:99-131.
[3]Zhou Y,Zhao J,Liu L.Meta-selective transition-metal catalyzed arene C—H Bond functionalization[J].Angew Chem Int Ed Engl,2009,48:7126-7128.
[4]Armin D M,Diederich F.Metal-catalyzed Cross-coupling Reactions[M].2nd Ed.Weinheim:Wiley-VCH,2004.
[5]Beletskaya I P,Cheprakov A V.Copper in cross-coupling reactions[J].The Post-Ullmann Chemistry Coord Chem Rev,2004,248:2337-2364.
[6]Masanari K,Daisuke N,Masahiro F,et al.Rh-catalyzed reductive coupling reaction of aldehydes with conjugated dienes promoted by triethylborane[J].Org Lett,2009,17:3794.
[7]徐伯华.2-碘代硒苯与2-唑烷酮在CuI催化下的C—N偶联反应机理的理论研究[J].原子与分子物理学报,2011,28(6):995.
[8]于海珠,傅尧,白小宇,等.亚铜催化的碳杂偶联反应机理[J].化学进展,2010,22(4):557.
[9]李来才,蔡皖飞,汪晓慧,等.N-(邻氯苯基)苯甲酰胺在CuX(X=I,Br)催化下分子内O-芳基化反应机理[J].物理化学学报,2009,25:2101.
[10]Ullmann F.The Ullmann condensation and the synthesis of diarylamines[J].Ber Dtsch Chem Ges,1903,36:2382-2384.
[11]Kohn W,Sham L J.Self-Consistent equations including exchange and correlation effects[J].Phys Rev,1965,A140:1133.
[12]Hariharan P C,Pople J A.The influence of polarization functions on molecular orbital hydrogenation energies[J].Theory Chem Acta,1973,28:213.
[13]Reed A E,Weinhold F,Curtiss L A,et al.Natural bond orbital a nalysis of molecular interactions:theoretical studies of binary complexes of HF,H2O,NH3,N2,O2,F2,CO and CO2with HF,H2O and NH3[J].Chem Phys,1986,84:5687-5706.
[14]Biegler K F,Schönbohm J,Derdau R,et al.AIM 2000[S].Ver 2.0.Hamilton:McMaster University,2002.
[15]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 09[S].Rev A.02.Pitsburgh:Gaussian,2009.
