紫外分光光度法测定藤黄药材中总藤黄酸含量
2014-06-19裴来良胡海霞王效山
纪 娟,裴来良,王 斌,程 卉,胡海霞,王效山,周 安
(1.安徽中医药大学科研实验中心,安徽 合肥 230038;2.安徽省中药研究与开发重点实验室,安徽 合肥 230031;3.安徽中医药大学药学院,安徽 合肥 230031)
紫外分光光度法测定藤黄药材中总藤黄酸含量
纪 娟1,2,裴来良2,3,王 斌1,2,程 卉1,胡海霞2,3,王效山2,3,周 安1,2
(1.安徽中医药大学科研实验中心,安徽 合肥 230038;2.安徽省中药研究与开发重点实验室,安徽 合肥 230031;3.安徽中医药大学药学院,安徽 合肥 230031)
目的 建立藤黄药材中总藤黄酸含量的测定方法。方法 采用紫外分光光度法,以新藤黄酸为对照品,在360nm处对藤黄样品中的总藤黄酸进行含量测定。结果 新藤黄酸浓度在5.304~26.520mg/L范围内,与吸收度的线性关系良好(r=0.999 5),平均加样回收率为98.53%(RSD=0.21%,n=9)。结论 该方法简便、准确、重现性好,可用于藤黄药材的质量控制。
藤黄药材;新藤黄酸;藤黄酸;紫外分光光度法
藤黄系藤黄科植物藤黄(Garciniahanburyi Hook.f.)所分泌出的干燥树脂,性寒、味酸、辛、涩、有毒,具破血散结、解毒、止血、杀虫之功效,用于治疗瘰疠、痈疸、疖肿等顽疾[1]。藤黄总酸制剂(包括注射剂和片剂)试用于临床上治疗多种恶性肿瘤,获得一定疗效[2]。藤黄中主要成分为藤黄酸、新藤黄酸为代表的桥环类化合物,迄今为止,已从藤黄药材中分离得到数十种桥环类化合物[3-4],该类化合物是藤黄抗肿瘤药理活性的物质基础。藤黄提取物及其单体成分藤黄酸(图1)、新藤黄酸(图2)具有显著的抗肿瘤活性[5-7],且毒性低。

图1 藤黄酸化学结构式 图2 新藤黄酸化学结构式
目前藤黄中桥环类化合物的分析方法主要有薄层色谱法[8]、高效液相色谱法[9]、液相色谱质谱联用法[10]等,薄层色谱法和高效液相色谱法在测定总藤黄酸时一般只针对一种或几种指标性成分定量,不能反映药材中总藤黄酸的含量。陈优生等[11]采用二氯氧锆显色后比色法测定总藤黄酸含量,然而显色反应易受温度和光照等诸多因素影响,造成稳定性不理想。为了有效地控制藤黄药材中总藤黄酸的质量,本实验拟根据藤黄中桥环类化合物在360nm左右均有紫外吸收特点,采用紫外分光光度法对藤黄中总藤黄酸进行了测定,为藤黄药材的质量控制及开发利用提供依据。
1 仪器与材料
UV-2550型紫外分光光度计:日本岛津公司;KQ-3000VDB超声波清洗器:江苏省昆山超声仪器有限公司;BP2110电子分析天平:德国Sartorius。
3批不同的藤黄药材由本课题组购于安徽亳州药材市场(批号分别为20091136、20100528、20100702),均经安徽中医药大学药学院彭华胜教授鉴定;新藤黄酸对照品由安徽中医药大学药学院药物研究所提取分离,其纯度经高效液相色谱面积归一化法测定其纯度为99.9%;其他试剂均为分析纯。
2 方法和结果
2.1 溶液的制备
2.1.1 新藤黄酸对照品储备液的制备:精密称取干燥至恒质量的新藤黄酸对照品26.52mg置于100 ml容量瓶中,用甲醇溶解并定容,即得265.2g/ml对照品储备液。
2.1.2 供试品溶液的制备:将藤黄药材粉碎,过40目筛,干燥至恒质量。取藤黄粉末30mg,精密称定,置于100ml容量瓶中,加甲醇溶解并稀释至刻度,超声提取10min,放冷至室温,补足甲醇至刻度。精密移取1ml置于10ml容量瓶中,甲醇定容至刻度,即得供试品溶液。
2.2 测定波长的选择 将“2.1.1”项下新藤黄酸对照品储备液用甲醇稀释10倍和“2.1.2”项下供试品溶液一起用紫外分光光度计进行扫描,测得对照品和供试品在200~500nm范围内的吸收光谱(见图3)。图3表明,藤黄药材和新藤黄酸对照品在360 nm波长处均有强吸收,故采用360nm处吸收波长测定总藤黄酸含量。
2.3 线性关系考察 精密量取265.2g/ml新藤黄酸对照品储备液10ml置100ml容量瓶中,加甲醇稀释至刻度。精密移取2.0、4.0、6.0、8.0、10.0ml稀释液于10ml容量瓶中,分别标记为1、2、3、4、5号溶液,用甲醇稀释至刻度,得浓度为5.30、10.61、15.91、21.22、26.52g/ml系列溶液。以甲醇为空白,测定各系列溶液在360nm下的吸光度。以新藤黄酸浓度(c)为横坐标,吸光度(A)为纵坐标,得线性回归方程为:A=0.024 88c-0.000 36,r=0.999 5(n=3)。结果表明,新藤黄酸对照品溶液吸收度与浓度在5.30~26.52g/ml范围内呈良好的线性关系。

图2 新藤黄酸对照品(A)和藤黄药材供试品(B)的紫外吸收光谱图
2.4 精密度测定 将“2.3”项下1、3、5号新藤黄酸对照品溶液作为低、中、高3个质量浓度,分别测其吸光度,各自连续测5次,考察仪器精密度,结果RSD值均低于0.4%,说明该方法精密度良好,符合含量分析要求。
2.5 稳定性测定 精密称取一批藤黄药材粉末10.44、19.96、30.40mg,按“2.1.2”项下方法操作得藤黄药材低、中、高3个质量浓度溶液,于室温中放置0、1、3、5、7、9h,分别测定吸光度,考察样品稳定性。见表1。结果表明藤黄供试品液在9h内稳定。
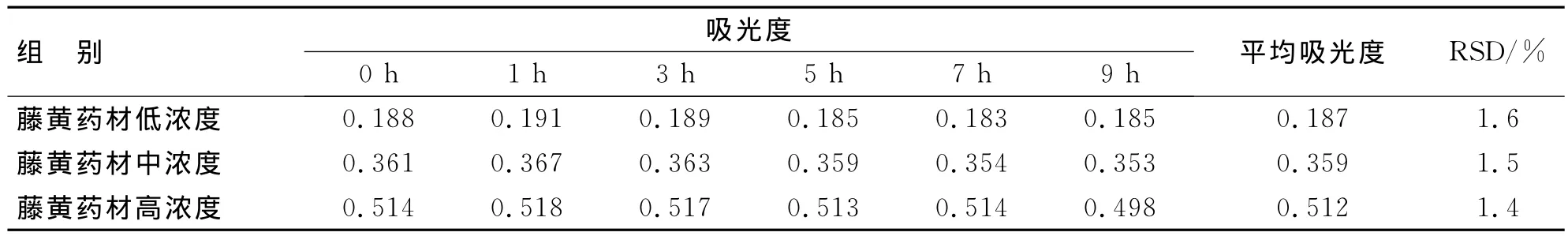
表1 藤黄供试品液稳定性测定结果
2.6 重复性测定 精密称取质量分别为30.86、29.68、29.60、29.04、30.28、29.82mg同一批藤黄药材(批号20100528)6份,按“2.1.2”项下同法制备样品溶液,在波长360nm处测定吸光度,按照“2.3”项线性方程计算总藤黄酸含量,考查重复性,结果平均含量为70.2%,RSD为1.6%(n=6)。结果表明本法重现性良好。
2.7 加样回收率测定 精密称取已知含量藤黄药材19.96mg,置于100ml容量瓶中,加甲醇溶解并稀释至刻度,超声提取10min,放冷至室温,补足甲醇至刻度。从中精密移取1.0ml溶液,同样移取9次,分别置于9个10ml容量瓶。将9个容量瓶平均分为3组,分别加入新藤黄酸对照品溶液(26.52 g/ml)4.5、5.5、6.5ml,加甲醇定容。分别测定吸光度,计算加样回收率,RSD值为0.21%。见表2。结果表明该方法具有较好的加样回收率。
2.8 含量测定 取3批藤黄药材(批号20091136)粉末各适量,分别按“2.1.2”项下方法制备供试品溶液,根据线性回归方程得出样品的总藤黄酸含量。见表3。结果表明3批藤黄药材的总藤黄酸含量相对较高。

表2 加样回收率测定结果(n=9)
3 讨论
在药材提取过程中,分别考察了提取5、10、15、20min对总藤黄酸含量提取的影响,发现提取10 min后含量变化不大,所以选择10min作为提取总藤黄酸的时间。早期藤黄药材质量控制研究多以薄层色谱及高效液相色谱为主,操作繁琐,选取指标往往难以代表整个藤黄药材中总藤黄酸含量。本课题组前期通过高效液相色谱二极管阵列检测器与电喷雾质谱联用分析了藤黄中桥环类化合物,获得了相应化合物的最大紫外吸收和分子量信息,结果表明藤黄中桥环类化合物在波长360nm处均有最大紫外吸收[10]。本实验根据该类化合物紫外吸收特点,采用紫外分光光度法建立了藤黄中总藤黄酸含量的测定方法,从实验结果来看,此方法准确可靠,结果稳定,可作为藤黄药材中总藤黄酸含量测定方法。

表3 藤黄样品中总藤黄酸含量测定(n=3)
[1]江苏新医学院《中药大辞典》编写组.中药大辞典:第2部分[M].上海:上海科学技术出版社,1997:564.
[2]藤黄抗癌研究协作组.藤黄(总体)抗癌实验与临床研究报告[J].江西医药,1982(3):1-5.
[3]Asano J,Chiba K,Tada M,et al.Cytotoxic xanthones fromGarciniahanburyi[J].Phytochemistry,1996,41(3):815-820.
[4]贾明美,寿清耀,谭青,等.藤黄化学成分的研究[J].化学学报,2008,66(22):2513-2517.
[5]Guo QL,Lin SS,You QD,et al.Inhibition of human telomerase reverse transcriptase gene expression by gambogic acid in human hepatoma SMMC-7721cells[J].Life Sci,2006,78(11):1238-1245.
[6]Li Q,Cheng H,Zhu G,et al.Gambogenic acid inhibits proliferation of A549cells through apoptosis-inducing and cell cycle arresting[J].Biol Pharm Bull,2010,33(3)415-420.
[7]雷秋模,刘金妹,龚德恩.藤黄抗癌的实验研究[J].中华肿瘤杂志,1985,7(4):282.
[8]叶定江,吴皓,胡勇,等.藤黄及其炮制品中藤黄酸含量的比较[J].中国中药杂志,1995,20(10):601-602.
[9]周安,李庆林,彭代银,等.HPLC法测定藤黄中藤黄酸和新藤黄酸的含量[J].中国中医药信息杂志,2008,15(8):53-54.
[10]周安,李庆林,彭代银,等.高效液相色谱-质谱联用法鉴定中药藤黄中桥环类化合物[J].药学学报,2008,43(8):838-842.
[11]陈优生,冯锋,尤启冬.ZrOCl2比色法测定藤黄中总藤黄酸的含量[J].中国药师,2002,5(12):731-732.
Determination of Total Gambogic Acid in Gamboge by Ultraviolet Spectrophotometry
JIJuan1,2,PEILai-liang2,3,WANGBin1,2,CHENGHui1,HUHai-xia2,3,WANGXiao-shan2,3,ZHOUAn1,2
(1.ExperimentalResearchCenter,AnhuiUniversityofChineseMedicine,AnhuiHefei230038,China;2.Key LaboratoryofChineseMedicineResearchandDevelopmentinAnhuiProvince,AnhuiHefei230031,China;
3.SchoolofPharmacy,AnhuiUniversityofChineseMedicine,AnhuiHefei230031,China)
Objective To establish a method for determining the content of total gambogic acid in gamboge.Methods With gambogenic acid as a reference substance,ultraviolet spectrophotometry was adopted to determine the content of total gambogic acid in gamboge sample at a wavelength of 360nm.Results The concentration of gambogenic acid showed a good linear relationship with optical density within 5.304-26.520mg/L (r=0.999 5),and the average recovery rate was 98.53% (RSD=0.21%,n=9).Conclusion The determination method is simple,accurate,and repeatable and can be used for the quality control of gamboge.
gamboge;gambogenic acid;gambogic acid;ultraviolet spectrophotometry
R927.2
A
10.3969/j.issn.2095-7246.2014.02.032
纪娟(1989-),女,硕士研究生
周安,anzhou0901@163.com
p
