1-苯基-1,2-丙二酮的振动光谱和理论计算研究
2014-05-25樊瑞雪薛佳丹万军民赵彦英
樊瑞雪,薛佳丹,万军民,赵彦英
(浙江理工大学理学院,杭州310018)
1-苯基-1,2-丙二酮的振动光谱和理论计算研究
樊瑞雪,薛佳丹,万军民,赵彦英
(浙江理工大学理学院,杭州310018)
获取1-苯基-1,2-丙二酮(PPD)在环己烷和乙腈溶剂中的电子吸收光谱,结合含时密度泛函理论(TDDFT)B3LYP-TD/6-311++G(d,p)计算,指认出A吸收带的跃迁主体为π→π*的跃迁。获取PPD A-带共5个不同激发波长下的共振拉曼光谱,并结合密度泛函理论(DFT)方法研究A吸收带的Franck-Condon区域结构动力学,其共振拉曼光谱被指认为Franck-Condon区域5个活性振动模的基频、倍频和组合频,其中的伸缩振动、C6C5C10/C7C8C9伸缩振动和环上CH的面内摇摆(v10、v11)对拉曼光谱强度贡献最大,表明PPD的S4激发态在Franck-Condon区域内的结构动力学主要沿这三个反应坐标展开。
1-苯基-1,2-丙二酮;激发态结构动力学;密度泛函理论计算;共振拉曼光谱
0 引 言
α-二羰基化合物特殊的分子结构赋予了它很多重要的应用,在生物化学领域可以用于蛋白质交联[1]、无酶交联[2]的Maillard反应,在有机合成中可以应用于合成新型环糊精衍生物[3]。α-二羰基化合物现在已经成为生物化学领域重要的研究对象。
α-二羰基化合物分子中存在分子内酮式-烯醇式之间的互变异构体[4-5],并且 ==O C-==C O在异构化过程中所呈现出来的扭角直接影响了这类分子的光化学性质[6-7]。在对PPD以及其他丙二酮类化合物的光谱学研究中,Arnett等[5]发现,当光激发丙二酮类分子时,具有平面型 ==O C-==C O构型的分子比非平面型分子更容易发光。研究者们得出结论,α-二羰基化合物中 ==O C-==C O存在扭角时,能量耗散途径中,S1态和T1态都有所减少。Quang Shen等[8]在气相条件下,通过电子衍射的实验方法得到PPD的分子结构,==O C-==C O的扭转角为129.9°,苯环几乎与它相邻的羰基共平面。
1-苯基-1,2-丙二酮(1-phenyl-1,2-propanedione,PPD)作为典型的α-二羰基化合物除了用于不对称有机合成反应外[9-10],还可用于光致聚合牙齿树脂复合材料的光敏剂[11]。Leonard等[12]研究了PPD分子n-π*的电子吸收光谱,发现扭转角度对PPD的吸收有很大影响。Susy Lopes等[13]研究了低温基质隔离下PPD的分子异构化以及紫外诱发的PPD去羰基化现象,进一步探讨了PPD的光化学和光物理现象。本文结合前人的研究成果,利用共振拉曼光谱技术结合密度泛函理论计算,研究了PPD的激发态结构动力学,通过分析振动光谱和电子吸收光谱,考察PPD在激发态时分子结构以及电子振动耦合吸收等方面的信息。
1 实验部分
1.1 试剂和仪器
环己烷(99.9%,光谱纯,TFDIA公司);乙腈(99.9%,光谱纯,TFDIA公司);1-苯基-1,2-丙二酮(PPD,98%,分析纯,东京化成工业株式会社)。
Cary 50型紫外可见分光光度计(瓦里安公司,美国);Nicolet Raman 960 FT-Raman光谱仪(尼高力公司,美国),Nicolet Avatar370 FT-IR光谱仪(尼高力公司,美国),共振拉曼光谱仪(自制)。
1.2 实验方法
分别以环己烷和乙腈做溶剂,配制PPD溶液的浓度约为3.00~4.00 mmol/L。共振拉曼光谱实验装置和方法参照文献[14]。通过四倍频激光线(波长266 nm)和氢气受激拉曼位移管获得共振拉曼光谱实验所用的激发波长。为保证实验过程中样品的新鲜性,样品通过蠕动泵输送。溶液放置于150 mL锥型瓶中,经导管在循环泵抽送下输送至喷嘴成液膜状流出,然后再经导管流回锥型瓶。拉曼信号的检测由CCD检测器完成。每个样品拉曼信号的检测时间为60~150 s,作为一次输出数据。采集20~30次(检测时间及采集次数由样品谱图的性噪比来确定),最后得到共振拉曼光谱。通过共振拉曼实验中获取的纯溶剂拉曼谱图,使用origin软件包完成对溶液共振拉曼光谱中溶剂峰的扣减,得到5个激发波长下PPD分别在环己烷和乙腈溶剂中的共振拉曼光谱。其中,共振拉曼光谱的自吸收校正和实验装置中信号收集系统的强度校正原理见文献[15],具体校正通过Origin软件的自编程序完成。
2 理论计算
本文所有量子化学计算均由Gaussian 09W程序包[16]完成。采用密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)方法,在B3LYP/6-311++ G(d,p)计算水平下获得PPD的几何结构和简正振动频率,在B3LYP-TD/6-311++G(d,p)计算水平得到PPD的电子跃迁能。
3 结果与讨论
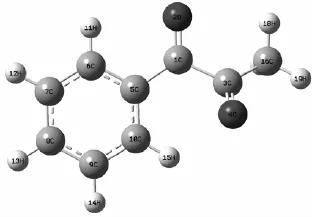
图1 PPD的几何构型(气相)
3.1 几何结构
计算研究了基态时1-苯基-1,2-丙二酮(PPD)的几何结构,如图1所示。在B3LYP/6-311++G(d,p)计算水平下PPD在基态时呈非平面型的分子结构的扭转角为135°,与文献[8]报道的结果基本一致,属于C1点群。
3.2 振动光谱
已有文献报道了PPD的分子结构[8]、光致异构化[13]等方面的信息,但在理论计算以及振动光谱的获取和指认等方面,尚无文献报道。为了进一步获取PPD的激发态结构动力学信息,以及对其共振拉曼光谱进行指认,在B3LYP/6-311++G(d,p)计算水平下得到了PPD在气相以及CPCM溶剂模型环己烷溶剂中的拉曼光谱,并通过实验测定了PPD的傅里叶变换拉曼(FT-Raman)光谱和傅里叶变换红外(FT-IR)光谱。图2为CPCM/C6H12和气相中PPD的计算拉曼光谱和实验拉曼光谱图,其中CPCM/C6H12是PPD在CPCM溶剂模型下环己烷溶剂中的计算拉曼光谱,gas phase是PPD在气相中的计算拉曼光谱,FT-Raman in solid是固体粉末PPD的FT-Raman实验光谱。通过对计算拉曼频率和实验所得傅立叶拉曼频率的对比,发现计算拉曼光谱与实验拉曼光谱吻合良好,说明常温下PPD主要是以非平面反式构型的形式存在,且溶剂中的计算光谱与气相中的计算光谱差别很小,说明在溶剂中PPD构型不变。根据PPD的FT-IR和FTRaman光谱的实验值,结合计算拉曼光谱,对PPD的振动光谱进行了归属和指认,结果见表1。表1列出了PPD计算所得的振动频率及其光谱指认和FT-IR,FT-Raman,共振拉曼光谱(R.Raman)实验的实验值。
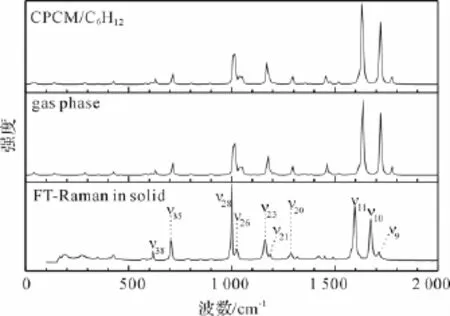
图2 PPD在CPCM/C6H12以及气相中计算拉曼光谱和实验拉曼光谱图
PPD的傅里叶拉曼光谱中,υ10(1 674 cm-1)是C1O2的伸缩振动;υ11(1 597 cm-1)是C6C5C10/ C7C8C9的伸缩振动+环上CH的面内弯曲;υ20(1 288 cm-1)是C3C1C5不对称伸缩振动+甲基伞式振动+环上CH的面内弯曲;υ23(1 161 cm-1)是C3C1C5对称伸缩振动+环上CH/甲基的面内弯曲;υ26(1 022 cm-1)是O4C3C1C16的面外变形+ C7C8C9对称伸缩;υ28(1 001 cm-1)是环上CH的面外弯曲+甲基面内弯曲;υ35(706 cm-1)是环上CH的面外弯曲+C5C1O2C3的面外变形+甲基的面内弯曲;υ38(617 cm-1)是环的变形振动。
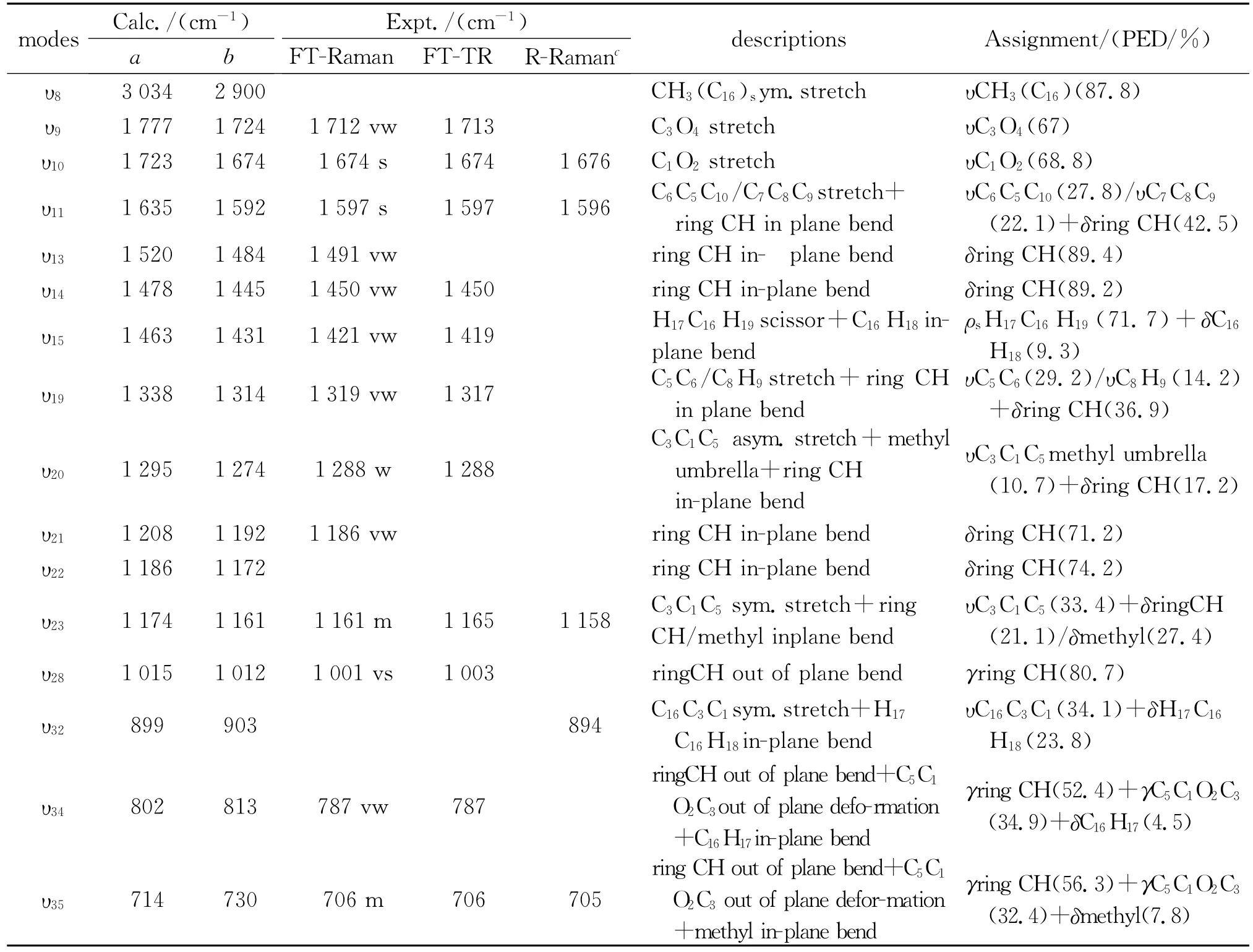
表1 在B3LYP/6-311+G(d,p)计算水平下PPD的振动光谱指认
3.3 电子光谱研究
图3是PPD在环己烷和乙腈溶剂中的紫外吸收光谱。共振拉曼光谱实验所选的激发波长如图中的箭头所示,分别为239.5、245.9、252.7、266 nm和273.9 nm。由图3可见,随着溶剂极性增大,吸收光谱发生了蓝移的现象,说明溶剂极性对PPD的电子跃迁产生了一定的影响。
表2是B3LYP-TD/6-311++G(d,p)计算水平下PPD的电子跃迁能(ΔE)和振子强度(f)。由表2数据可见,在200~400 nm紫外光谱区域,计算得到的气相振子强度值f=0.190 8,与环己烷溶剂中的实验值(f=0.104 6)稍有差别,说明在PPD的激发态电子跃迁中,存在溶剂效应的影响。
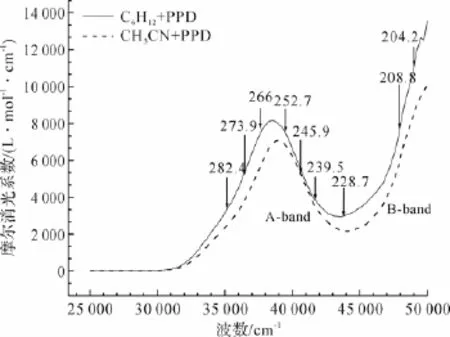
图3 PPD在环己烷和乙腈中的紫外吸收光谱

表2 在气相条件下计算得到的PPD电子跃迁能(△E)和振子强度(f)
表2中,气相计算条件下,PPD最高占有分子轨道(HOMO,轨道39)是氧原子上的孤对电子所形成的非键轨道n,第二(HOMO-1,轨道38)和第三最高占有分子轨道(HOMO-2,轨道37)是π成键轨道πH-1和πH-2。第一(LUMO,轨道40)、第二(LUMO+1,轨道41)和第三最低空价键轨道(LUMO+ 2,轨道42)均为π*反键轨道,π*L、π*L+1、π*L+2。其中276 nm附近的吸收带被指认为是πH-2→π*L和πH-1→π*L和πH-1→π*L+1的跃迁引起的。前线分子轨道跃迁如图4所示。
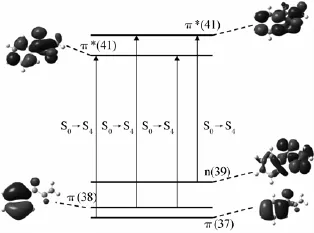
图4 PPD电子跃迁分子轨道图
3.4 共振拉曼光谱
PPD在环己烷和乙腈溶剂中不同激发波长下的共振拉曼光谱示于图5。图6示出了PPD在环己烷溶剂中波长245.9 nm和266 nm下的共振拉曼光谱及其光谱指认。
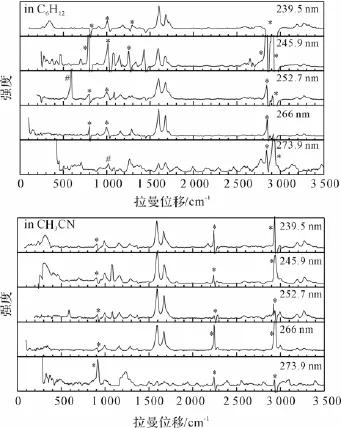
图5 不同激发波长下PPD在环己烷和乙腈溶剂中的共振拉曼光谱
在图5中,239.5、245.9、252.7、266 nm和273.9 nm波长下共振拉曼光谱对应于电子吸收光谱中的A-带吸收,其强度模式主要隐含了S4态激发态结构动力学信息。
由图6可知,A-带的共振拉曼光谱可以被指认为Franck-Condon区域中5个活性振动模的基频、倍频和组合频:v10(C1O2的伸缩振动)、v11(C6C5C10/C7C8C9伸缩振动+环上CH的面内摇摆)、v23(C3C1C5对称伸缩振动+环上CH/甲基的面内摇摆)、v32(C16C3C1对称伸缩振动+H17C16H18面内摇摆)、v35(环上CH的面外弯曲+C5C1O2C3面外变形+甲基面内弯曲)。其中v10(C1O2的伸缩振动)和v11(C6C5C10/C7C8C9伸缩振动+环上CH的面内摇摆)对激发态结构起决定作用,它们的基频、倍频和组合频占据了共振拉曼光谱强度的绝大部分,这说明PPD的S4(态激发态结构动力学主要沿的伸缩振动、环上的伸缩振动和环上的面内摇摆等反应坐标展开。
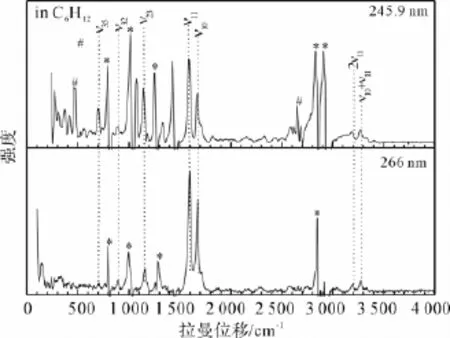
图6 245.9 nm和266 nm下PPD在环己烷溶剂中共振拉曼光谱的指认
4 结 论
本文采用密度泛函理论方法,结合共振拉曼光谱技术研究了1-苯基1,2-丙二酮(PPD)的A带电子激发和Franck-Condon区域结构动力学,得出如下结论:
a)气相条件下,PPD的稳定构型是非平面型的,属于C1点群。理论计算的优化结果和傅里叶拉曼实验的结果的对比证实了这一点。
b)PPD的电子吸收光谱表明,其电子吸收的跃迁主体是π→π*的跃迁,归属为A-电子吸收带,该吸收带对S4态Franck-Condon区域激发态结构有主要作用。
[1]Miller A G,Meade SJ,Gerrard J A.New insights into protein crosslinking via the Maillard reaction:structural requirements,the effect on enzyme function,and predicted efficacy of crosslinking inhibitors as anti-ageing therapeutics[J].Bioorganic&Medicinal Chemistry,2003,11(6):843-852.
[2]Meade SJ,Miller A G,Gerrard J A.The role of dicarbonyl compounds in non-enzymatic crosslinking:a structure-activity study[J].Bioorganic&Medicinal Chemistry,2003,11(6):853-862.
[3]Kida T,Michinobu T,Zhang W,et al.A facile synthesis of novel types of cyclodextrin derivatives by insertion of an aromatic dicarbonyl spacer into a permethylatedαcyclodextrin skeleton[J].Chemical Communications,2002(15):1596-1597.
[4]Pawlikowski M,Zgierski M Z,Orlandi G.Absorption and emission spectra of molecules subject to hindered internal rotation with an application toα-dicarbonyls[J]. Chemical Physics Letters,1984,105(6):612-616.
[5]Arnett JF,Newkome G,Mattice W L,et al.Fxcited electronic states of the alpha.dicarbonyls[J].Journal of the A-merican Chemical Society,1974,96(14):4385-4392.
[6]Gómez-Zavaglia A,Fausto R.Matrix-isolation and solid state low temperature FT-IR study of 2,3-butanedione(diacetyl)[J].Journal of Molecular Structure,2003,661:195-208.
[7]Lopes S,Gómez-Zavaglia A,Lapinski L,et al.Matrixisolation FTIR spectroscopy of benzil:probing the flexibility of the CC torsional coordinate[J].The Journal of Physical Chemistry A,2004,108(40):8256-8263.
[8]Shen Q,Hagen K.Molecular structures and conformations of phenylglyoxal and 1-phenyl-1,2-propanedione as determined by gas-phase electron diffraction[J].The Journal of Physical Chemistry,1993,97(5):985-988.
[9]Toukoniitty F,Mäki-Arvela P,Kuusisto J,et al.Solvent effects in enantioselective hydrogenation of 1-phenyl-1,2-propanedione[J].Journal of Molecular Catalysis A:Chemical,2003,192(1):135-151.
[10]Mikkola J P,Toukoniitty B,Toukoniitty F,et al. Utilisation of on-line acoustic irradiation as a means to counter-effect catalyst deactivation in heterogeneous catalysis[J].Ultrasonics Sonochemistry,2004,11(3):233-239.
[11]Schroeder W,Arenas G,Vallo C.Monomer conversion in a light-cured dental resin containing 1-phenyl-1,2-propanedione photosensitizer[J].Polymer International,2007,56(9):1099-1105.
[12]Leonard N J,Mader P M.The influence of steric configuration on the ultraviolet absorption of 1,2-diketones[J].Journal of the American Chemical Society,1950,72(12):5388-5397.
[13]Lopes S,Gómez-Zavaglia A,Lapinski L,et al.Conformational flexibility,UV-induced decarbonylation,and FTIR spectra of 1-phenyl-1,2 propanedione in solid xenon and in the low temperature amorphous phase[J].The Journal of Physical Chemistry A,2005,109(25):5560-5570.
[14]Jian J W,Zhang H B,Chen C Q,et al.Resonance Raman spectroscopic and density functional theory investigation of the excited state structural dynamics of 2-mercapto-1-methylimidazole[J].Journal of Raman Spectroscopy,2013,44(4):582-589.
[15]许宗平,赵彦英,王惠钢,等.N-甲基吡咯-2-甲醛激发态结构动力学及其溶剂效应的共振拉曼光谱和密度泛函理论研究[J].物理化学学报,2012,28(1):62-72.
[16]Foresman JB,Frisch M J.GAUSSIAN 03[M].Pittsburgh PA:Gaussian Inc,2003.
Study on VibrationaI Spectrum and TheoreticaIInvestigation of 1-PhenyI-1,2-Propanedione
FAN Rui-xue,XUE Jia-dan,WAN Jun-min,ZHAOYan-ying
(School of Science,Zhejiang Sci-Tech University,Hangzhou 310018,China)
Flectronic absorption spectrum of 1-phenyl-1,2-propanedione(PPD)was gained in cyclohexane and acetonitrile solvent.In combination of Time-dependent Density Functional Theory(TD-DFT)B3LYP-TD/6-311++G(d,p),transition subject of absorption band A was identified to beπ→π*.Resonance Raman spectra under 5 different excitation wavelengths of PPD Band A were obtained.Structural dynamics of Franck-Condon region of absorption band A was studied in combination of DFT.The resonance spectra were assigned to five fundamental frequencies,frequency multiplication and combined frequencies in Franck-Condon region,where C1=O2stretching vibration,C6C5C10/C7C8C9stretching vibration and in-plane rockingυ10,υ11)on the ring CH had the largest contributions to Raman spectrum intensity.This indicates structural dynamics of PPD in S4excited state mainly spreads along the three reaction coordinates.
1-phenyl-1,2-propanedione;structural dynamics in excited state;density functional theory;resonance Raman spectra
O643.12
A
(责任编辑:许惠儿)
1673-3851(2014)04-0481-06
2013-12-12
国家自然科学基金(21273202);浙江省自然科学基金(LY13B030009)
樊瑞雪(1989-),女,河南安阳人,硕士研究生,研究方向为光化学反应动力学。
赵彦英,F-mail:zhaoyying2008@hotmail.com
