2-吡嗪腈的激发态结构动力学研究
2014-05-25邱朦朦薛佳丹王惠钢
邱朦朦,薛佳丹,王惠钢
(浙江理工大学理学院,杭州310018)
2-吡嗪腈的激发态结构动力学研究
邱朦朦,薛佳丹,王惠钢
(浙江理工大学理学院,杭州310018)
采用密度泛函理论计算获得了2-吡嗪腈(2-CP)在气相中的电子吸收光谱,通过实验获得了2-CP在水、甲醇、乙腈和环己烷中的紫外吸收光谱和共振拉曼光谱。在B3LYP/6-311+G(d,p)计算水平上,发现A-带和B-带的电子跃迁主体为π→π*。A-带共振拉曼光谱指认为6个基频的振动模式及其泛频和组合频,其中υ16(N1/N4面内弯曲振动),υ14(吡嗪环呼吸振动),υ10(C2C7伸缩振动+C3H9面内弯曲振动)的基频、泛频和组合频对拉曼光谱强度的贡献最大。在B-带,2-CP的结构反应动力学主要沿着υ5(C2C3/C5C6对称伸缩振动)和υ14(吡嗪环呼吸振动)反应坐标展开。A-带与B-带的主要贡献振动模的强度不同,结果表明,A-带与B-带的激发态反应动力学的结构存在差异。
2-吡嗪腈;紫外光谱;电子跃迁;共振拉曼光谱;密度泛函理论;光诱导反应动力学
0 引 言
吡嗪及烷基吡嗪等吡嗪衍生物是一种重要的化学中间体,同时也是合成食品香料、农用化学品、药品、抗静电剂、纺织织物整理液等的重要原料及中间体[1]。吡嗪类衍生物可以应用于咖啡香、肉香等具有特殊风味的食品添加剂[2]。2-吡嗪腈(2-CP)是一种非常重要的化工产品,它还可以用作医药中间体,经过水解反应可以制备一线抗结核药物吡嗪酰胺等医药产品[3-4]。它在药物化学、生物化学、催化化学和能源化学等[5-7]的发展中起着重要的作用。
吡嗪是许多重要杂环化合物的母核,其衍生物的电子激发态在核酸UV光损伤中扮演着重要的角色。因此吡嗪及其衍生物的光物理化学性质,尤其是激发态结构动力学的研究成为大家关注的焦点。Guo[8]通过对吡嗪、嘧啶分子之间的光物理和光化学异构化反应的研究,发现锥形交叉点CI(S1/ S0)是一个关键的中间点,将CI(S2/S1)和过渡态TS连接起来。Su[9]通过研究吡嗪的光致异构化过程,发现从吡嗪的第二激发态S2(1B2u,ππ*)开始,在吡嗪和嘧啶的势能面之间经过一个锥形交叉通道,最后又回到嘧啶的基态势能面上。2-吡嗪腈作为吡嗪的衍生物,其振动光谱及激发态诱导反应动力学鲜有人关注。本文采用密度泛函理论计算方法[10-11]研究2-CP的紫外吸收光谱、电子跃迁、振动光谱和共振拉曼光谱及其指认,为进一步研究2-CP及其相关物质的光诱导激发态反应动力学提供基础。
1 实验部分
1.1 实验药品及仪器
1.1.1 实验药品
2-吡嗪腈(2-cyanopyrazine),其分子式C5H6N2,相对分子质量94.12,优级纯,98%(北京百灵威科技有限公司);
环己烷(cyclohexane),其分子式C6H12,相对分子质量84.16,色谱纯,99.9%(Honeywell公司);
乙腈(acetonitrile),其分子式CH3CN,相对分子质量41.01,色谱纯,99.9%(Fulltime公司);
甲醇(methanol),其分子式CH3OH,相对分子质量32.04,色谱纯,99.9%(TFDIA公司);
高纯水(water),其分子式H2O,相对分子质量18.00,分析纯,99.0%(杭州娃哈哈集团有限公司)。
1.1.2 实验仪器
Varian Cary 50 CONC紫外可见光谱仪(驭锘实业有限公司,中国),Nicolet Raman 960 FT-Raman光谱仪(尼高力公司,美国),Nicolet Avatar370 FT-IR光谱仪(尼高力公司,美国),共振拉曼光谱仪(自组装仪器)。
1.2 实验方法
用环己烷、乙腈、甲醇和水做溶剂,2-吡嗪腈的浓度根据不同的激发波长调整样品浓度,以便获得更强的共振拉曼信号。共振拉曼的实验方法见参考文献[12]。首先,根据紫外吸收光谱中不同的电子吸收带,选择合适的激发波长,所用激发波长由四倍频激光线及其H2受激拉曼位移管获得。为保证样品不被激光照射所破坏,液样采用循环流动的方式。溶液放置于150 mL锥型瓶中,经导管在循环泵抽送下输送至喷嘴成液膜状流出,然后再经导管流回锥型瓶。将120~180 s搜集到的拉曼信号作为一次输出数据,将20~35次得到数据累计叠加后得到相应的共振拉曼光谱。采用改编后origin3.5软件扣减掉光谱中溶剂的拉曼峰,再用origin5.0扣除基线得到样品在该溶剂中的共振拉曼光谱。
1.3 理论计算
本文采用密度泛函理论方法,所有的量子化学计算均由Gaussian 09W程序包[13]完成。在B3LYP/6-311+G(d,p)计算水平下获得2-吡嗪腈(2-CP)的几何结构优化及振动频率,在B3LYPTD/6-311+G(d,p)计算水平下获得2-CP的电子跃迁能。
2 结果与讨论
2.1 几何结构
2-吡嗪腈(2-CP)的几何结构如图1所示,在B3LYP-TD/6-311+G(d,p)计算水平下,得到优化后的几何结构,其中,2-CP主要是由离域的吡嗪环和氰基组成。从图中还可看到,2-CP的结构高度对称,所有的原子位于同一平面内,而且只有一个对称面,经过计算后得到2-CP的几何结构属于Cs点群。
2.2 振动光谱分析
为了对共振拉曼光谱进行指认和激发态结构动力学研究,笔者在B3LYP/6-311+G(d,p)水平下计算获得了2-吡嗪腈的振动频率和对称信息,结合其振动频率的红外和拉曼活性与傅立叶拉曼光谱进行对比,结果如图2所示。再通过Gussian View软件对每个频率的振动情况观察,并对每个振动频率进行了指认。通过计算的拉曼频率和实验得到的傅立叶拉曼频率对比,进行了二次校正,发现计算值和理论值较一致。根据在B3LYP/6-311+G(d,p)计算水平下的振动频率,并结合FT-Raman、FTIR和共振拉曼光谱的实验值,对2-CP的振动光谱进行了详细指认,见表1。从振动光谱结合计算的光谱指认来看,在100~1 700 cm-1范围内,可以观察到有19个拉曼活性模,可以很明显地发现在这19个拉曼活性模中属于A′的有14个;有16个红外活性模,其中在这16个红外活性模中属于A′的有12个。
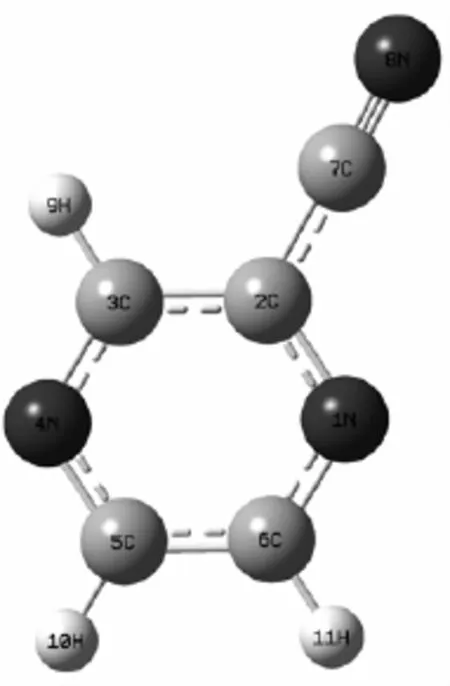
图1 2-吡嗪腈的几何结构
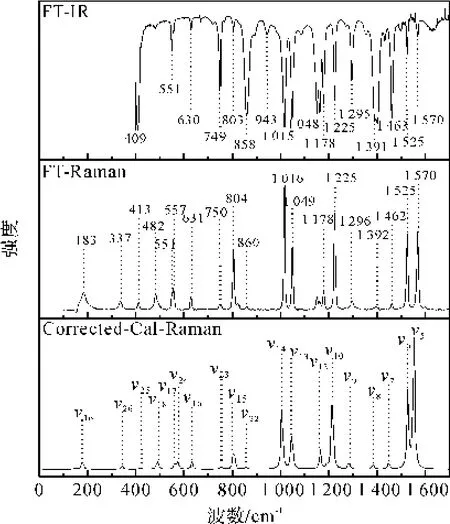
图2 2-CP的FT-IR、FT-Raman以及校正后的计算拉曼光谱的对比

表1 在B3LYP/6-311+G(d,p)计算水平下2-CP的振动光谱指认
2.3 电子光谱分析
图3是2-吡嗪腈在水、甲醇、乙腈和环己烷溶液中的紫外吸收光谱,共振拉曼光谱实验所选的激发波长如图中的箭头所示,它们分别是217.8、228.7、239.5、252.7、266.0、273.9 nm和282.4 nm。由图3得出两个电子吸收带,分别是230~290 nm称之为A-带,230 nm之前的吸收带称为B-带。运用朗伯-比尔定律的数学表达式A=ε·b·c,计算获得2-吡嗪腈在环己烷、乙腈、甲醇和水溶剂中跃迁A-带的摩尔消光系数ε分别为7 775.0、8 363.3、8 921.8、10 266.9 L/(mol·cm);跃迁B-带的摩尔消光系数ε分别为9 070.3、10 133.0、11 500.5、11 888.0 L/(mol·cm)。
表2是B3LYP-TD/6-311+G(d,p)计算水平下获得的2-吡嗪腈的电子跃迁能(ΔE)和振子强度(f)。表2计算结果显示,计算得出两个允许电子跃迁带245 nm(f=0.170 4)和213 nm(f=0.161 5),与其所对应的实验值269 nm(f=0.111 1)和210 nm(f= 0.178 0)强吸收带基本相一致。在不同溶液中,其紫外吸收光谱的最大吸收峰较相似,所以不同溶剂对其A-带和B-带的电子跃迁没有较大影响。

图3 2-CP在水、甲醇、乙腈和环己烷中的紫外光谱
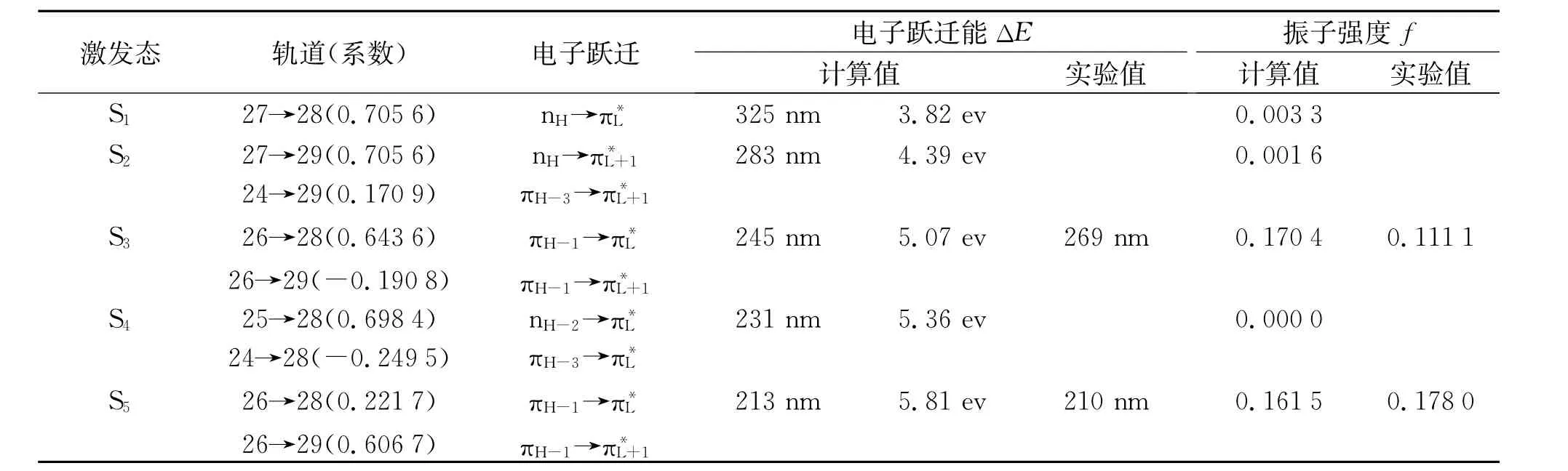
表2 B3LYP-TD/6-311+G(d,p)计算获得的2-吡嗪腈的电子跃迁能(△E)和振子强度(f)
图4所示为2-吡嗪腈的主要电子跃迁轨道。2-吡嗪腈所涉及的电子跃迁轨道24、25、26、27、28、29,如图4所示,在269 nm附近的A-吸收带所涉及的主要电子跃迁轨道是24、26、28和29,根据休克尔法则,图中轨道24(HOMO-3)和26(HOMO-1)的主特征是吡嗪环的π成键,轨道28(LUMO)和29(LUMO+1)的主特征是吡嗪环的π*反键,而轨道25(HOMO-2)和26(HOMO)的主特征是n轨道。将2-吡嗪腈在269nm(A-带)附近的电子吸收(0.64(26→28跃迁)指认为π→π*跃迁。在210 nm附近的B-吸收带所涉及的主要跃迁轨道是26和29,其电子吸收(0.61(26→29跃迁)也可以指认为π→π*跃迁。由表2中轨道系数及振子强度数据可得,2-吡嗪腈在269 nm附近的A-吸收带为πH-1→跃迁,B-吸收带为πH-1→的跃迁。振子强度数据表明,πH-1→和πH-1→的跃迁是紫外吸收的主体。而暗态nH-2→的跃迁对总振子强度来说可以忽略不计。对于2-吡嗪腈激发态分子的初始电子运动来说,具有离域特征。

图4 2-吡嗪腈的主要电子跃迁轨道
2.4 共振拉曼光谱分析
通过共振拉曼实验,选取相应的共振激发波长,对2-吡嗪腈(2-CP)在不同溶剂中共振拉曼谱图进行了研究。图5是在266.0 nm(最大吸收处)激发波长下,2-CP在水、甲醇、乙腈和环己烷溶剂中的共振拉曼光谱谱峰指认。图5对2-CP在环己烷溶剂中的共振拉曼光谱的基频及泛频进行了详细指认。从图5看出,A-带的共振拉曼光谱可被指认为6个Franck-Condon区域的活性振动模的基频。即υ16(628 cm-1)/N1/N4面内弯曲振动,υ15(804 cm-1)/吡嗪环的呼吸振动,υ14(1 012 cm-1)/吡嗪环的呼吸振动,υ13(1 046 cm-1)/C3H9/C5H10/C6H11面内弯曲振动,υ10(1 222 cm-1)/C2C7伸缩振动+C3H9面内弯曲振动,υ7(1 442 cm-1)/C3H9/C5H10/ C6H11面内弯曲振动。

图5 在266.0 nm激发波长下,2-CP在环己烷、乙腈、甲醇和水溶剂中的共振拉曼光谱图

图6 在217.8 nm激发波长下,2-CP在乙腈、甲醇和水中的共振拉曼光谱指认(0~3 500 cm-1)
实验获取了在B-带的217.8 nm激发波长下,2-CP在乙腈、甲醇和水中的共振拉曼光谱,如图6所示,并对其共振拉曼光谱的谱峰归属进行了指认。该共振拉曼光谱振动模主要体现了B-带电子跃迁时的反应动力学信息。通过与A-带的活性振动模对比,发现基本一致,但其共振拉曼光谱谱峰的相对强度有很大的区别。其中v12完全消失,而振动模v5和v14在其拉曼谱峰中的贡献较大,v5的强度最大,v14次之,这充分说明在217.8 nm激发波长下,反映了2-CP的B-带激发态反应动力学的部分特征。υ5(C2C3/C5C6对称伸缩振动)的相对强度在B-带明显强于A-带,可能是由于高激发态的预共振增强效应所造成的。A-带与B-带的主要贡献振动模不同,表明A-带与B-带的激发态反应动力学结构存在着差异。在B-带,2-CP的结构反应动力学主要沿着υ5(C2C3/C5C6对称伸缩振动)、υ14(吡嗪环呼吸振动)反应坐标。
图7展示了2-CP的水溶液在217.8、228.7、239.5、252.7、266.0、273.9 nm和282.4 nm激发波长下的共振拉曼光谱指认。选择282.4、273.9、266.0 nm和252.7 nm作为A-带的的激发波长,它所反映的是A-带的电子跃迁信息,选择217.8 nm作为B-带的激发波长同时所反应的是B-带的电子跃迁信息。而239.5 nm和228.7 nm作为检测可能的预共振从很高的激发态到252.7 nm。通过图中的比较,我们得知共振拉曼光谱在不同的激发波长下很相似,但其中振动模的强度相差较大。最显著的是,A-带强度最大的振动模是v14和v10,而B-带强度最大的振动模是v5。A-带中的v7、v10、v13、v14、v15和v16模的强度都较强,而对于B-带中只有v5和v14模的强度较强,从活性模的强度及数量上我们可以得出A-带结构动力学与B-带结构动力学明显不同。
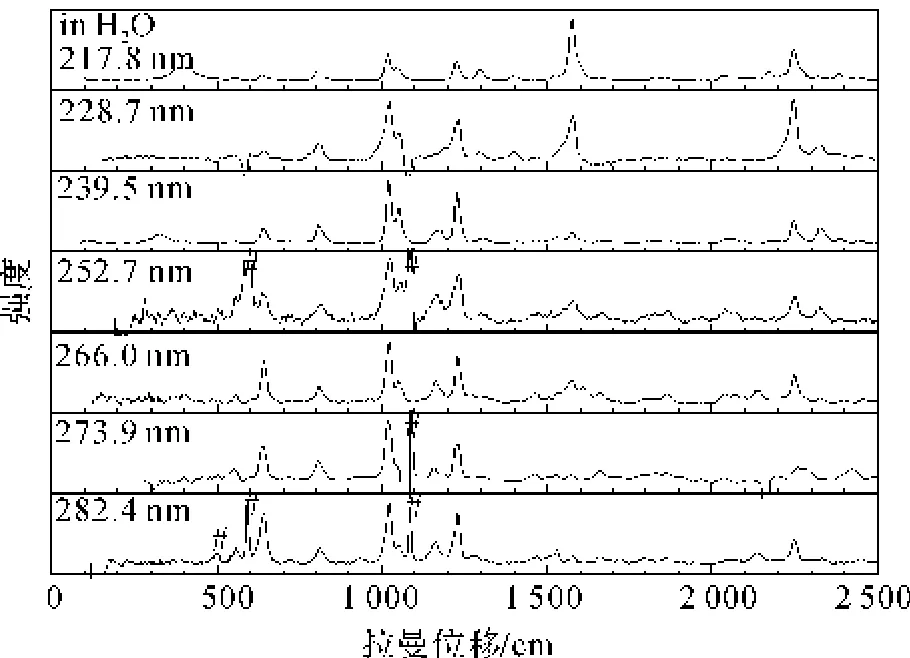
图7 2-CP在水中不同波长下的共振拉曼光谱(光谱已做了强度校正和溶剂扣减,#为激光线位置)
3 结 论
采用密度泛函理论,在B3LYP/6-311+G(d,p)计算水平下,解释了2-CP的电子吸收光谱、振动光谱以及共振拉曼光谱,得到如下结论:
a)2-吡嗪腈(2-CP)主要是由离域的吡嗪环和氰基组成。2-CP的所有的原子位于同一平面内,且只有一个对称面,在B3LYP-TD/6-311+G(d,p)计算水平下得到2-CP的几何结构属于Cs点群;
b)2-CP的紫外吸收光谱图在非质子性溶剂(环己烷和乙腈)和质子性溶剂(水和甲醇)中,它们的谱带形状及最大吸收峰处无明显差异,这说明溶剂对其电子跃迁没有明显的影响,A-吸收带(269 nm)明态为πH-1→和πH-3→跃迁,暗态为nH-2→;B-吸收带(210 nm)为πH-1→和πH-1→跃迁;
c)2-CP的A-带结构反应动力学主要沿着υ16(N1/N4面内弯曲振动),υ14(吡嗪环呼吸振动),υ10(C2C7伸缩振动+C3H9面内弯曲振动)反应坐标,B-带与A-带的主要贡献振动模的强度不同,表明A-带与B-带的激发态反应动力学的结构存在差异。在B-带,2-CP的结构反应动力学主要沿着υ5(C2C3/C5C6对称伸缩振动)、υ14(吡嗪环呼吸振动)反应坐标。
[1]Anand R,Jyothi T M,Rao B S.A comparative study on the catalytic activity of ZnO modified zeolites in the synthesis of alkylpyrazines[J].Applied Catalysis A:General,2001,208(1):203-211.
[2]卫舒平,张 华,徐晓彬,等.吡嗪衍生物的天然存及其在食用香精中的应用[J].香料香精化妆品,2000(2):25-31.
[3]Subrahmanyam M,Kulkarni S J,Rao A V R.Catalyst preparation studies for the synthesis of 2-methylpyrazine[J].Indian Journal of Chemical Technology,1995,2(5):237-240.
[4]OpletalováV,Patel A,Boulton M,et al.5-Alkyl-2-pyrazinecarboxamides,5-alkyl-2-pyrazinecarbonitriles and 5-alkyl-2-acetylpyrazines as synthetic intermediates for antiinflammatory agents[J].Collection of Czechoslovak Chemical Communications,1996,61(7):1093-1101.
[5]Wieser M,Heinzmann K,Kiener A.Bioconversion of 2-cyanopyrazine to 5-hydroxypyrazine-2-carboxylic acid with Agrobacterium sp.DSM 6336[J].Applied Microbiology and Biotechnology,1997,48(2):174-176.
[6]Srilakshmi C,Lingaiah N,Suryanarayana I,et al.In situ synthesis of ammonium salt of 12-molybdophosphoric acid on iron phosphate and the ammoxidation functionality of the catalyst in the transformation of 2-methylpyrazine to 2-cyanopyrazine[J].Applied Catalysis A:General,2005,296(1):54-62.
[7]Fried L F,Manaa M R,Pagoria P F,et al.Design and synthesis of energetic materials 1[J].Annual Review of Materials Research,2001,31(1):291-321.
[8]Guo J L,Liu C,Xie B B,et al.Vibronic coupling and excited-state reaction dynamics of pyrazine in 1 1B2u(1ππ*)state by resonance Raman spectroscopy and CASSCF calculation[J].Journal of Raman Spectroscopy,2012,43(10):1477-1486.
[9]Su M D.CASCSF study on the photochemical transposition reactions of pyrazines[J].The Journal of Physical Chemistry A,2006,110(30):9420-9428.
[10]Parr R G,Yang W.Density-functional Theory of Atoms and Molecules[M].London:Oxford University press,1989,285-317.
[11]Jones R O,Gunnarsson O.The density functional formalism,its applications and prospects[J].Reviews of Modern Physics,1989,61(3):689.
[12]Wang L B,Zhang W,Shen S,et al.The excited state dynamics study of di-2-pyridylketone in the A-band and B-band absorptions by using resonance Raman spectroscopy,IR and UV-visible spectroscopy[J].Journal of Raman Spectroscopy,2012,43(10):1465-1471.
[13]Liu M X,Xie B B,Li M J,et al.A-band structural dynamics of thioanisole by resonance Raman spectroscopy[J].Journal of Raman Spectroscopy,2013,44(3):440-446.
Study on StructuraI Dynamics of 2-Cyanopyrazine in Excited State
QIU Meng-meng,XUE Jia-dan,WANG Hui-gang
(School of Science,Zhejiang Sci-Tech University,Hangzhou 310018,China)
The electronic absorption spectrum of 2-cyanopyrazine(2-CP)in gaseous phase was gained through Density Functional Theory.The UV absorption spectrum and resonance Raman spectrum of 2-CP were obtained experimentally in water,methanol,acetonitrile and cyclohexane solvent respectively.Based on B3LYP/6-311+G(d,p)calculation,it is found that electronic transition subject of Band A and Band B isπ→π*.Resonance Raman spectrum of Band A could be identified as vibration modes of 6 fundamental frequencies as well as their overtones and combined frequencies,where fundamental frequencies,overtonesand combined frequencies ofυ16(N1/N4 in-plane bending vibration),υ14(pyrazine ring breathing vibration)andυ10(C2C7 stretching vibration+C3H9 in-plane bending vibration)contribute mostly to Raman spectrum intensity.In Band B,structural reaction dynamics of 2-CP mainly spreads along reaction coordinates ofυ5(C2C3/C5C6 symmetrical stretching vibration)andυ14(pyrazine ring breathing vibration).The intensity of the main vibration models of Band A and Band Bis different.The result shows that,the structures of reaction dynamics of Band A and Band B in excited state are different.
2-cyanopyrazine;UV spectrum;electronic transition;resonance Raman spectrum;density functional theory;photoinduced state reaction dynamics
O643.12
A
(责任编辑:许惠儿)
1673-3851(2014)04-0474-07
2013-11-29
邱朦朦(1989-),男,安徽颍上人,硕士研究生,主要从事光化学反应动力学研究。
