RP-HPLC法同时测定不同产地金银花中木犀草苷和 6 种有机酸
2014-04-11李向阳屠万倩
李向阳, 屠万倩
(1.河南省食品药品检验所, 河南 郑州 450003; 2.河南省中医药研究院, 河南 郑州 450004)
RP-HPLC法同时测定不同产地金银花中木犀草苷和 6 种有机酸
李向阳1, 屠万倩2*
(1.河南省食品药品检验所, 河南 郑州 450003; 2.河南省中医药研究院, 河南 郑州 450004)
目的 建立反相高效液相色谱法测定不同产地金银花中木犀草苷、绿原酸、隐绿原酸、新绿原酸和异绿原酸A、 B、 C的方法。 方法 Phenomenex ODSC18色谱柱 (4.6 mm×250 mm, 5 μm), 以乙腈 (A)-0.4%磷酸 (B) 为流动相, 梯度洗脱 (0 ~10 m in, 5%→9%A; 10 ~30 min, 9%A; 30 ~60 min, 9% →30%A; 60 ~80 min, 30%A) ,体积流量 1.0 mL/min, 最大吸收波长下检测 (木犀草苷在 350 nm, 隐绿原酸、 绿原酸和新绿原酸在 327 nm, 异绿原酸 A、 B、 C在 330 nm)。 结果 7 种待测成分分离度良好; 各成分质量浓度与峰面积在测定范围内均呈良好线性关系(r≥0.999 0), 木犀草苷、 绿原酸、 隐绿原酸、 新绿原酸和异绿原酸 A、 B、 C的平均回收率 (RSD) 分别为 99.52% (2.56%)、 97.93%(2.19%)、 96.12%(1.19%)、 102.50%(1.63%)、 97.57%(1.60%)、 97.12%(2.64%)、100.32%(2.89%)。 结论 该方法简便、 准确、 重复性好, 可用于金银花的质量控制。
金银花; 木犀草苷; 有机酸; RP-HPLC
金银 花 为 忍 冬 科 植 物 忍 冬 Lonicera japonica 的花蕾, 具有清热解毒、 疏散风热的功效[1]。 金银花在我国河南、河北、山东等多地分布广泛,其中河南金银花种植历史悠久、产量较大,是金银花的道地产地和主产区,河南密县主产的金银花被习称为密银花或南 银花[2-3]。 近年来, 随金银花 的市场需求量增大,新产区不断发展,山东沂蒙山区(包括平邑、费县等地) 已成为除河南封丘、 新密等传统产区外新兴的金银花的重点生产基地,河南、山东两省主产的金银花产销量可达全国的近70%,成为国内金银花的主要来源[4]。
金银花中主要含有黄酮及苷类、环烯醚萜苷类和有机酸类成分[5]。 金银花中的黄酮类成分主要有木犀草素、 木犀草苷 (木犀草素-7-O-葡萄糖苷)等, 有机酸类成分主要除绿原酸 (5-O-咖啡酰基奎宁酸) 外, 还含有 4,5-O-二咖啡酰基奎宁酸 (异绿原酸 C)、 3,4-O-二咖啡酰基奎宁酸 ( 异绿原酸B)、 3,5-O-二咖啡酰基奎宁酸 (异绿原酸 A)、 1,3-O-二咖啡酰基奎宁酸 (洋蓟素)、 4-咖啡酰基奎宁酸 (隐绿原酸)、5-咖啡酰基奎宁酸 ( 新绿原酸) 等咖啡酰基奎宁酸[6-7]。 根据其化学结 构, 绿原酸与隐绿原酸、新绿原酸结构较为相似,异绿原酸 A、B、 C为同分异构化合物。 据报道,从金银花中提取检测的绿原酸、木犀草苷、异绿原酸A、B、C等多种成分具有清除自由基、 抗氧化作用[8]。
在现行 2010 年 版 《 中 国 药典》中, 采 用HPLC法分别测定金银花中绿原酸和木犀草苷的量, 除利用先进的仪器分析技术 (如 HPLC-DAD/ ESI-MS 联用测定金银花中 24 种黄酮、 环烯醚萜苷类和皂苷类成分) 的研究报道[9]外, 目前国内金银花的质量研究文献报道多集中在绿原酸和木犀草苷的定量 测 定 及 指 纹 图 谱 上[10-13]。 考 虑 到有 机 酸为金银花中的重要活性成分,且组成较复杂,采用较常见的反相高效液相色谱-二极管阵列检测器(HPLC-DAD), 本实验同时测定河南和山东主产的金银花药材中木犀草苷、绿原酸、隐绿原酸、新绿原酸和异绿原酸 A、 B、 C, 有助于全面评价金银花药材质量。
1 仪器试剂与药品
1.1 仪器 美国 Waters高效液相色谱仪 (2695 溶剂管理系统, 2996 二极管阵列检测器, 美国 Waters柱温箱, Empower2 工作站); ASS150 超声波提取器 (济宁科特超声电子有限责任公司 GT-350W); LIBROR-160DPT万 分之一分析天平 ( 日本岛津); AE240 十万分之一分析天平 (瑞士 METTLER公司)。
1.2 试剂与药品 金银花样品分别购自河南封丘、河南新密、河南郑州、山东平邑、山东临沂等金银花主产区,经河南省食品药品检验所雷留成副主任药师鉴定, 均 为 忍 冬科 植 物 忍冬 L.japonica 的 花蕾。绿原酸对照品 (中国药品生物制品检定所,含量测定用, 批号 110753-200413), 木犀草苷对照品 (中国药品生物制品检定所, 含量测定用,批号 111720-200604), 隐绿原酸、 新绿原酸、异绿原酸A、 异绿原酸 B和异绿原酸 C对照品 (购自天津马克生物技术有限公司),甲醇、乙腈为色谱纯 (德国 Merck 公司), 水为超纯水,其余试剂均为分析纯。
2 方法与结果
2.1 色 谱 条 件 Phenomenex ODS C18色 谱 柱(4.6 mm×250 mm,5 μm); 柱温 40 ℃; 流动相为乙腈-0.4%磷酸水溶液系统, 梯度洗脱 (0 ~10 min, 5% → 9%A; 10 ~30 min, 9%A; 30 ~60 min, 9%→30%A; 60 ~80 min, 30%A); 体积流量 1.0mL/min;进样量 10 μL; 绿原酸、隐绿原酸和新绿原酸检测波长 327 nm,木犀草苷 350 nm,异绿原酸 A、 异绿原酸 B和异绿原酸 C 330 nm。在此条件下,色谱图见图1。
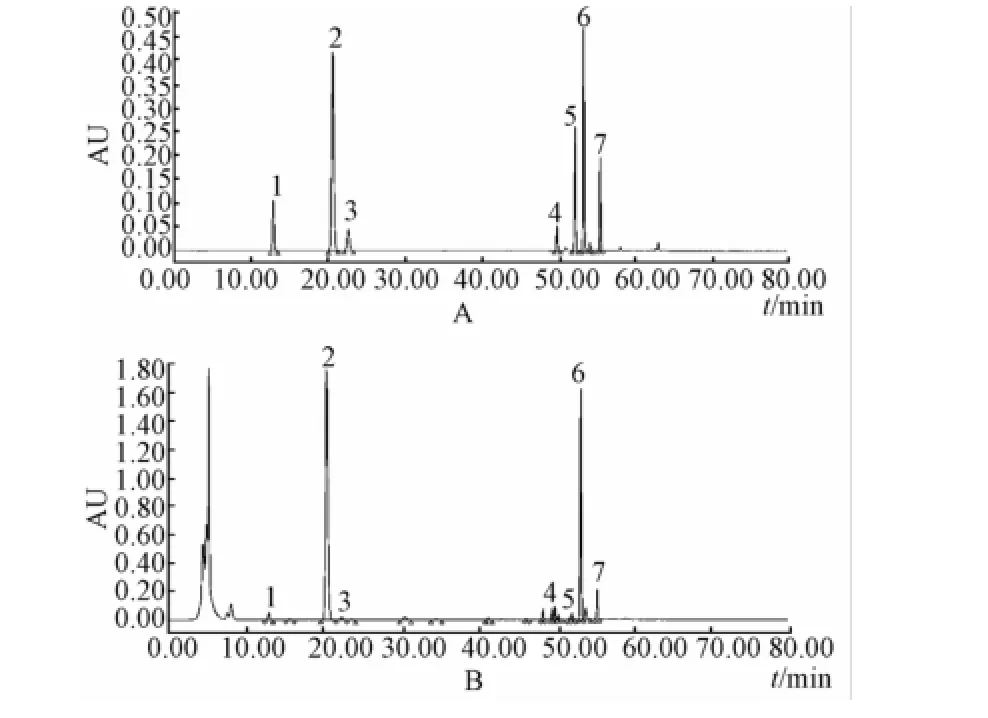
图1 对照品 (A) 与金银花样品 (B)HPLC色谱图Fig.1 HPLC chromatogram s of reference substances ( A) and sam p le of Lonicerae japonicae Flos( B)
2.2 对照品溶液的制备 精密称取木犀草苷、 隐绿原酸、绿原酸、新绿原酸、异绿原酸B、异绿原酸 A和异绿原酸 C对照品, 加 70%乙醇制成每 1 mL分别含木犀草苷 37.0 μg、隐绿原酸 48.0 μg、绿原酸 0.836 mg、 新绿原酸 48.8 μg、 异绿原酸 B 19.5 μg、 异绿原酸 A 0.254 mg和异绿原酸 C 55.6 μg的混合溶液, 作为对照品溶液。
2.3 供试品溶液的制备 取本品 2.0 g, 精密称定, 置具塞三角瓶中, 精密加入 70%乙醇 25 mL,称定质量, 超声处理1 h, 取出, 放冷, 再称定质量, 用 70%乙醇补足损失的质量, 摇匀, 滤过,续滤液用 0.45 μm微孔滤膜滤过, 即得。
2.4 供试品溶液的制备方法研究
2.4.1 提取溶剂的选择 取金银花样品 3 份, 各2.0 g, 精密称定, 分别精密加入甲醇、 70%乙醇和 50%乙醇 25 m L, 称定质量, 超声处理 1 h, 制成供试品溶液,测定供试品中木犀草苷、隐绿原酸、绿原酸、新绿原酸、异绿原酸B、异绿原酸A和异绿原酸C的量, 结果见表1。
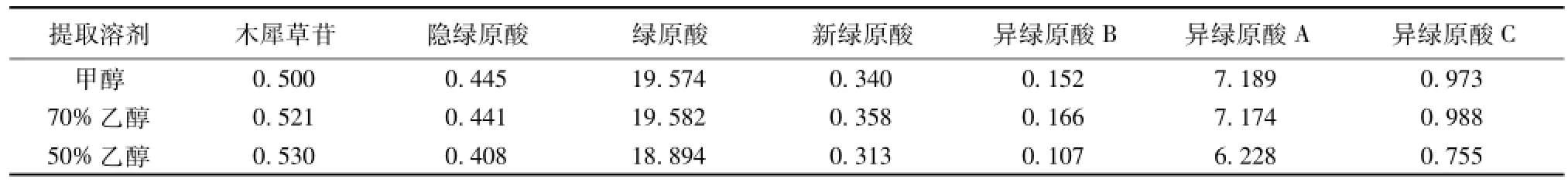
表 1 提取溶剂的选择 (mg·g-1)Tab.1 Selection of extraction solvents( mg·g-1)
2.4.2 提取方式的比较 取金银花样品 6 份,各2.0 g, 精密称定, 精密加入 70%乙醇 25 mL, 称定质量, 分别加热回流 30 min、 1 h、 2 h、 超声处理 30 min、 1 h、 2 h, 制成供试品溶液, 测定供试品中木犀草苷、隐绿原酸、绿原酸、新绿原酸、异绿原酸B、异绿原酸 A和异绿原酸 C的量, 结果见表2。

表 2 提取方式的比较 (mg·g-1)Tab.2 Com parison of extraction methods( mg·g-1)
2.5 线性关系考察 精密吸取 “2.2” 项下的对照品溶液 2、 5、 10、 15、20、 25 μL,分别注入液相色谱仪,测定。以峰面积对质量浓度进行线性回归, 回归方程线性范围及相关系数, 见表3。

表3 回归方程、线性关系及相关系数Tab.3 Regression equations, linear ranges and correlation coefficients
2.6 检测限和定量限的测定 取混合对照品溶液,按浓度稀释法进样分析 (分别按 10、 100、 1000、10 000 倍稀释), 测定色谱系统基线噪音。 以信噪比 3 ∶1 为检测限, 信噪比 10 ∶l为定量限。 经检测, 木犀草苷检测限为 0.9 ng, 定量限为 3.7 ng;隐绿原酸检测限为 1.2 ng, 定量限为 4.8 ng; 绿原酸检测限为 0.2 ng, 定量限为 0.8 ng; 新绿原酸检测限为 0.1 ng, 定量限为0.5 ng; 异绿原酸 B检测限为0.1 ng, 定量限为 0.2 ng; 异绿原酸 A检测限为 1.3 ng, 定量限为 5.1 ng; 异绿原酸 C检测限为0.7 ng,定量限为 2.8 ng。
2.7 精密度试验 吸取同一供试品溶液 10 μL,注入高效液相色谱仪,连续重复进样6次,测定各待测成分的峰面积并计算 RSD, 结果木犀草苷、隐绿原酸、绿原酸、新绿原酸、异绿原酸 B、 异绿原酸 A和异绿原酸 C的 RSD分别为 1.38%、2.62%、 1.07%、 2.42%、 2.57%、 1.28% 和1.85%。 结果表明, 本方法精密度较好。
2.8 稳定性试验 取同一供试品溶液, 分别于 0、2、 4、6、 8、 10 h 注入液相色谱仪, 测定各待测成分的峰面积并计算 RSD值, 结果木犀草苷、 隐绿原酸、绿原酸、新绿原酸、异绿原酸B、异绿原酸A和异绿原酸 C的 RSD分别为 2.05%、 2.86%、1.74%、 2.33%、 2.06%、1.71% 和 2.00%。 表明供试品溶液在10 h内稳定性较好。
2.9 重复性试验 取同一批金银花药材, 平行制备 6 份供试品溶液, 按 “2.3”项下方法制备成供试品溶液,按上述色谱条件进行 HPLC分析。 根据所得待测成分的峰面积计算其在样品中的含有量。结果显示:木犀草苷、隐绿原酸、绿原酸、新绿原酸、异绿原酸 B、 异绿原酸 A和异绿原酸 C的平均质量分数分别为0.523、 0.434、 19.610、 0.366、0.161、 7.305 和 0.966 mg/g, RSD 值 分 别 为1.48%、 2.97%、 1.64%、 1.98%、 2.69%、2.52%和 2.83%, 表明该方法重复性较好。
2.10 加样回收率试验 取已测定的金银花药材(木犀草苷、隐绿原酸、 绿原酸、新绿原酸、异绿原酸B、异绿原酸 A和异绿原酸 C的平均质量分数分别为 0.523、0.434、 19.610、0.366、 0.161、7.305 和 0.966 mg/g), 取约 1g,精密称定, 置具塞三角瓶中,分别精密加入木犀草苷、隐绿原酸、绿原酸、新绿原酸、异绿原酸B、异绿原酸A和异绿原酸 C对照品的混合 70%乙醇溶液 (质量浓度分别为 0.019 6 mg/mL、 0.019 0 mg/mL、 0.748 mg/mL、 0.016 8 mg/m L、 0.278 mg/mL、 0.045 mg/mL)25 mL, 以下按 “2.3” 项下操作, 平行操作,制得6份加样供试品溶液。吸取加样供试品溶液10μL, 注入液相色谱仪,测定木犀草苷、 隐绿原酸、绿原酸、新绿原酸、异绿原酸B、异绿原酸A和异绿原酸C的量。根据测得量和加入量,计算各待测成分的加样回收率, 结果见表4。

表4 加样回收率试验结果Tab.4 Results of recovery tests
2.11 样品测定 取不同产地的金 银 花, 按“2.3”项方法制备供试品溶液,测定样品中木犀草苷、隐绿原酸、绿原酸、新绿原酸、异绿原酸B、异绿原酸 A和异绿原酸 C的量, 平行测定 3次, 并计算,结果见表5。
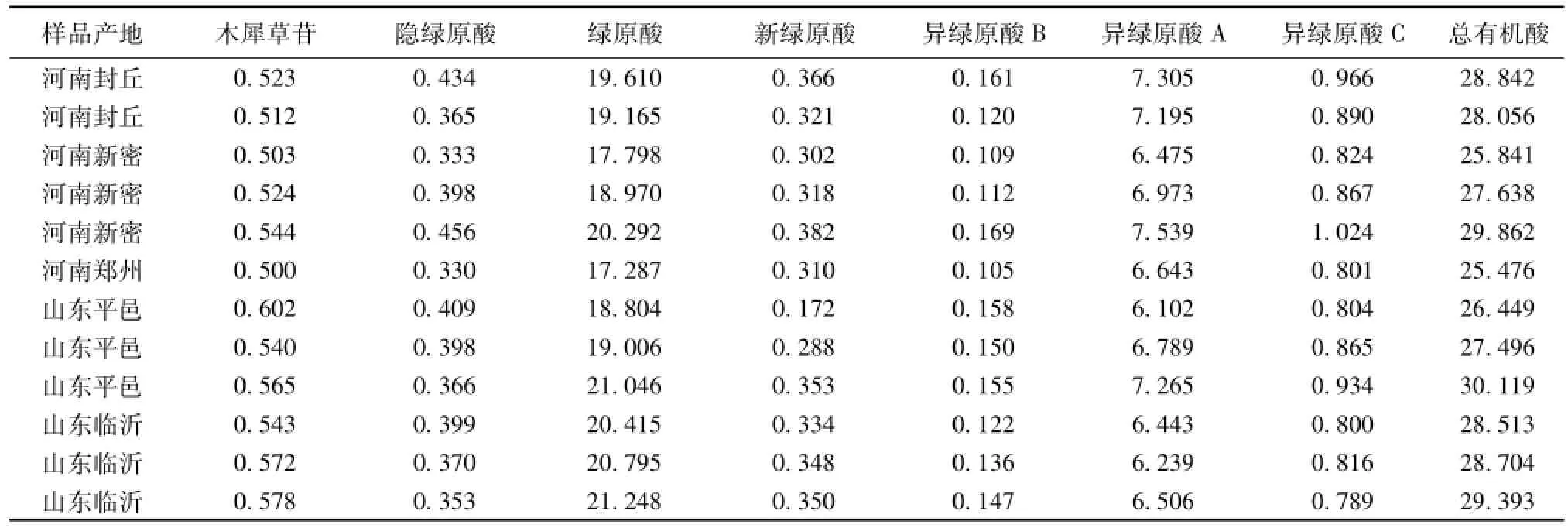
表 5 不同产地金银花样品的测定结果 (mg·g-1, n=3)Tab.5 Con tent determ ination of sam p les from differen t habitats(m g·g-1, n=3)
3 讨论
3.1 供试品溶液的制备 在实验中, 根据待测成分的理化性质,对供试品的提取溶剂 (甲醇、70%乙醇和50%乙醇)、 提取方式 (超声处理和加热回流)及提取时间等进行了考察和比较,确定了定量测定的供试品溶液的制备方法为取金银花2.0 g, 加 25 mL70%乙醇, 超声提取 1 h。
3.2 流动相的选择 金银花中木犀草苷、 绿原酸与异绿原酸类化合物化学结构和极性差异较大,难以采用固定的流动相同时测定,多用梯度洗脱方法实现待 测 成 分 的 分 离 和 检 测[14-15], 本 实 验 参 照《中国药典》 2010 年版一部金银花药材项下方法反复试验,确定了梯度洗脱程序,使待测成分峰形基本对称,与相邻峰可达较好的分离度,不同产地的药材样品均能准确测定。
3.3 测定波长的确定 在实验中,分别对木犀草苷、隐绿原酸、绿原酸、新绿原酸、异绿原酸B、异绿原酸A和异绿原酸C色谱峰进行吸收光谱扫描, 结果木犀草苷在 348 nm、 隐绿原酸、绿原酸和新绿原酸等咖啡酰基奎宁酸类成分色谱峰在 327 nm处、 异绿原酸 B、 异绿原酸 A和异绿原酸 C等二咖啡酰基奎宁酸色谱峰在329 nm处有最大吸收,参照 《中国药典》 2010 年版一部中木犀草苷的测定波长为 350 nm、 绿原酸的测定波长为 327 nm的有关规定, 选择 327 nm作为隐绿原酸、 绿原酸和新绿原酸的测定波长, 350 nm作为木犀草苷的测定波长, 330 nm作为异绿原酸 B、 异绿原酸 A和异绿原酸 C的检测波长。采用 Waters Empower II色谱工作站的定时波长功能, 分别以 45 min 和 51 min 为检测波长转换点, 检测波长依次由 327 nm变为350 nm及330 nm, 在同一色谱中同时检测金银花中的木犀草苷、隐绿原酸、绿原酸、新绿原酸、 异绿原酸 B、 异绿原酸 A和异绿原酸 C, 提高检测的灵敏度和准确性。此法不仅可以用于二极管阵列检测器,也可用于普通的多波长紫外检测器,具有普遍适用性,方便快速。
4 小结
在 《中国药典》 2010 年版一部金银花项下, 采用 HPLC法分别测定了绿原酸和木犀草苷的量,并规定金银花中绿原酸的量不得低于规定 1.5%(15 mg/g), 木犀草苷不得低于 0.05%(0.5 mg/g)[1]。在研究过程中,收集了河南和山东等金银花主产区的12 份样品, 实验结果表明, 12 份金银花样品中绿原酸和木犀草苷的量均符合药典规定。
同时,从收集到的样品测定结果来看,来自主产地的12份金银花样本中木犀草苷、绿原酸和 6种有机酸总量均无显著性的差异 (RSD值分别为5.89%、 6.46%和 5.40%),只是在各成分含有量构成上有所不同。排除隐绿原酸、新绿原酸和异绿原酸B等含有量较低、测定结果波动范围较大的成分,根据实验结果,观察产地对药材中化学成分的影响,结果提示产自山东的金银花样品中总有机酸、 绿原酸和木犀草苷的平均含有量 (28.446、20.219 和 0.567 mg/g) 略高于产自河南的样品(27.619、18.854 和 0.518 mg/g),而异绿原酸 A和异绿原酸 C的量则产自河南的样品 (7.022、8.95 mg/g) 略高于产自山东的样品 (6.557、 8.35 mg/g)。 有关金银花中化学成分的含有量差异与不同产区是否存在相关性,有待今后收集更多样品进行实验和比较。
考虑到金银花中含有多种异绿原酸成分,且异绿原酸A、异绿原酸C等成分在药材中含有量并不低,仅以绿原酸的量作为质控指标显然不够科学,建议进行多指标成分的定量测定来控制金银花及其制剂的内在质量。
[ 1 ] 国家药典委员会.中华人民共和国药典:2010 年版一部[S].北京: 中国医药科技出版社, 2010:205-206.
[2] 周凤琴,李 佳,冉 蓉,等.我国金银花主产区种质资源调查[J].现代中药研究与实践, 2010, 24(3):21-25.
[ 3 ] 杨俊杰, 李 娟.河南金银花产地生产状况调查[J].时珍国医国药, 2011, 22(4):974-975.
[4] 周凤琴,张永清,张 芳,等.山东金银花种质资源的调查研究[J].山东中医杂志, 2006, 25(4):268-271.
[ 5 ] 何 晶.金银花的化学成分及药理作用[J].天津药学,2007, 19(5):66-68.
[6] 刘 雄,高建德.金银花质量控制、化学成分及药理学研究进展[J].甘肃中医学院学报, 2006, 23(4):46-49.
[ 7 ] 贺 伟.金银花的化学成分及药理作用研究[J].中国医药导报, 2007, 4(24):8-9.
[ 8 ] Tang Dan, LiHuiun, Chen Jun, etal.Rapid and simplemethod for screening of natural antioxidants from Chinese herb Flos Lonicerae Japonicae by DPPH-HPLC-DAD-TOF/MS[ J] .J Sep Sci, 2008, 31(20):3519-3526.
[ 9 ] Chen Jun, Song Yue, Li Ping.Capillary high-performance liquid chromatography withmass spectrometry for simultaneous determination ofmajor flavonoids, iridoid glucosides and saponins in Flos Lonicerae[ J] .J Chromatogr A, 2007, 1157(1/2): 217-226.
[10] 李红霞, 王雪芹, 李振国, 等.不同产地金银花与山银花主要成分的含量比较[J].中国药房, 2011, 22(31): 2935-2937.
[11] 周凤琴, 冉 蓉, 李 佳, 等.山东 10 个不同种质金银花中绿原酸含量及其品质评价[J].山东中医杂志, 2007, 26 (7):478-480.
[12] 冯文宇, 田 吉, 何 兵.济银花与川银花绿原酸含量及提取率实验研究[J].中药材, 2006, 29(1):30-31.
[13] 容 蓉, 闫 斌, 吕青涛, 等.不同产地金银花药材的HPCE指纹图谱分析[J].化学分析计量, 2006, 15(6): 66-68.
[14] 张 璐, 曾文雪.HPLC-ELSD同时测定山金银花中绿原酸、 木犀草苷和川续断皂苷乙的含量[J].江西中医学院学报, 2011, 23(4):40-42.
[15] 王荣梅, 徐丽华, 林永强.法同时测定银黄含片中 6 个咖啡酰奎宁酸类成分的含量[ J].药物分析杂志, 2012, 32 (1):57-60.
Sim ultaneous determ ination of galuteolin, and six organic acids in Lonicerae Japonicae Flos from different habitats by RP-HPLC
LIXiang-yang1, TUWan-qian2*
(1.Henan Provincial Instituteof Food and Drug Control, Zhengzhou450003, China; 2.Henan Provincial Academy of Traditional ChineseMedicine, Zhengzhou 450004, China)
AIM To establish a RP-HPLC method for simultaneously determining the levels of galuteolin,chlorogenic acid,cryptochlorogenic acid, neochlorogenic acid, isochlorogenic acid A, B, and C in Lonicerae Japonicae Flos from different habitats.M ETHODS A Phenomenex ODSC18column(4.6 mm×250 mm, 5 μm) was used with the mobile phase consisting of acetonitrile(A)-0.4%phosphoric acid(B)in gradient elution mode(0-10 min, 5%A→9%A; 10-30 min,9%A;30-60 min, 9%A→30%A; 60-80 min, 30%A) ,at the flow rate of 1.0 mL/min.The UV detection wavelength was setat themaximum absorption wavelength, 350 nm for galuteolin, 327 nm for chlorogenic acid, cryptochlorogenic acid and neochlorogenic acid, 330 nm for isochlorogenic acid A, B, and C.RESULTS Excellent chromatographic separation was achieved with good linearity (r≥0.999 0)within the studied concerntration ranges.The average recovery rates(RSD)of galuteolin, chlorogenic acid, cryptochlorogenic acid,neochlorogenic acid, isochlorogenic acid A, B, and C were 99.52% (2.56%), 97.93%(2.19%), 96.12%(1.19%), 102.50%(1.63%), 97.57%(1.60%), 97.12% (2.64%), 100.32%(2.89%), respectively.CONCLUSION Themethod is simple, accurate and reproducible.It can be used for the quality control of Lonicerae Japonicae Flos.
Lonicerae Japonicae Flos; galuteolin; organic acid; RP-HPLC
R284.1
:A
:1001-1528(2014)02-0353-06
10.3969/j.issn.1001-1528.2014.02.030
2013-04-07
河南省重点科技攻关计划资助项目 (112102310023)
李向阳 (1968—) , 男, 副 主 任 药 师, 从 事 中 药 质 量 控 制 研 究 与 药 品 检 验 工 作。 Tel:(0371)63388283, Email:lixia632 @126.com
*通信作者: 屠万倩 (1970—), 女, 副研究员, 研究方向: 中药分析。 Tel:(0371)66331718
