积雪草酸衍生物的合成及抗肿瘤活性的研究
2014-03-25孟艳秋
薛 菁, 王 楠, 孟艳秋
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
积雪草酸(Asiatic acid,AA),又名亚细亚酸,为五环三萜类化合物,具有乌苏烷型骨架,存在于龙脑香科植物龙脑香的树脂和挥发油中,也可由伞形科植物草中的积雪草苷水解而得.近代研究表明,积雪草酸具有广泛的药理活性,如抗肿瘤[1-2]、抗氧化[3]、神经保护[4]、抗抑郁[5]、保护肝脏[6]和抗炎[7]等.本文以积雪草酸(化合物1)为原料,保留五环三萜骨架,对其2位、3位、12位、23位和28位进行结构修饰,设计并合成了12个积雪草酸类似物.
1 实验部分
1.1 仪器与试剂
化合物熔点在X-4数字显示显微熔点测定仪上测定,温度未经校正.红外光谱在ThermoNicolet 470FT红外光谱仪上测定(KBr压片).质谱在菲尼根hermo-finnigan LCQ equipment仪上测定.核磁共振氢谱在Bruker ARX-300型核磁共振分析仪上测定,CDCl3为溶剂,TMS为内标.薄层色谱采用高效色谱硅胶GF254(青岛海洋化工厂生产).柱色谱用200~300目柱色谱硅胶(青岛海洋化工厂生产).流动相采用石油醚(沸程在60~90 ℃)和乙酸乙酯的混合溶液.所有试剂均为分析纯或化学纯,部分试剂使用时根据需要进一步纯化.
1.2 化学合成
1.2.1 2-乙酰氧基-3,23-O-异亚丙基-乌苏烷型-12-烯-28-羧酸 (2a)的合成
合成路线如图1所示.将100.0 mg(0.20 mmol)化合物1和对甲基苯磺酸17.0 mg(0.20 mmol)溶解在DMF中,然后滴加2,2-二甲氧基丙烷0.5 mL(2.0 mmol),在室温下搅拌,TLC检测反应,反应完毕,用质量分数5 %的NaOH调节pH至7~8,混合物用乙酸乙酯提取,有机层用蒸馏水及饱和NaCl溶液洗涤,无水MgSO4干燥,抽滤,减压蒸馏得白色固体(化合物2),mp:156.4~158.2 ℃.IR(KBr):3 400,1 698,1 200 cm-1.将产物用THF溶解,加入少量的DMAP,滴加乙酸酐10 mmol,室温下反应并用TLC检测反应的终点(显色剂:体积分数95 %的硫酸乙醇溶液,展开剂为V(石油醚)V(乙酸乙酯)=3/1).反应结束后,蒸干溶剂,充分水洗,过滤,滤饼洗至中性.自然晾干,得化合物(2a)96 mg,总收率:42.1 %.IR(KBr),cm-1:2 951,2 923,1 747,1 697,1 457,1 370,1 235,1 045.ESI-MS:1 227(2M+2CH3CO)+.
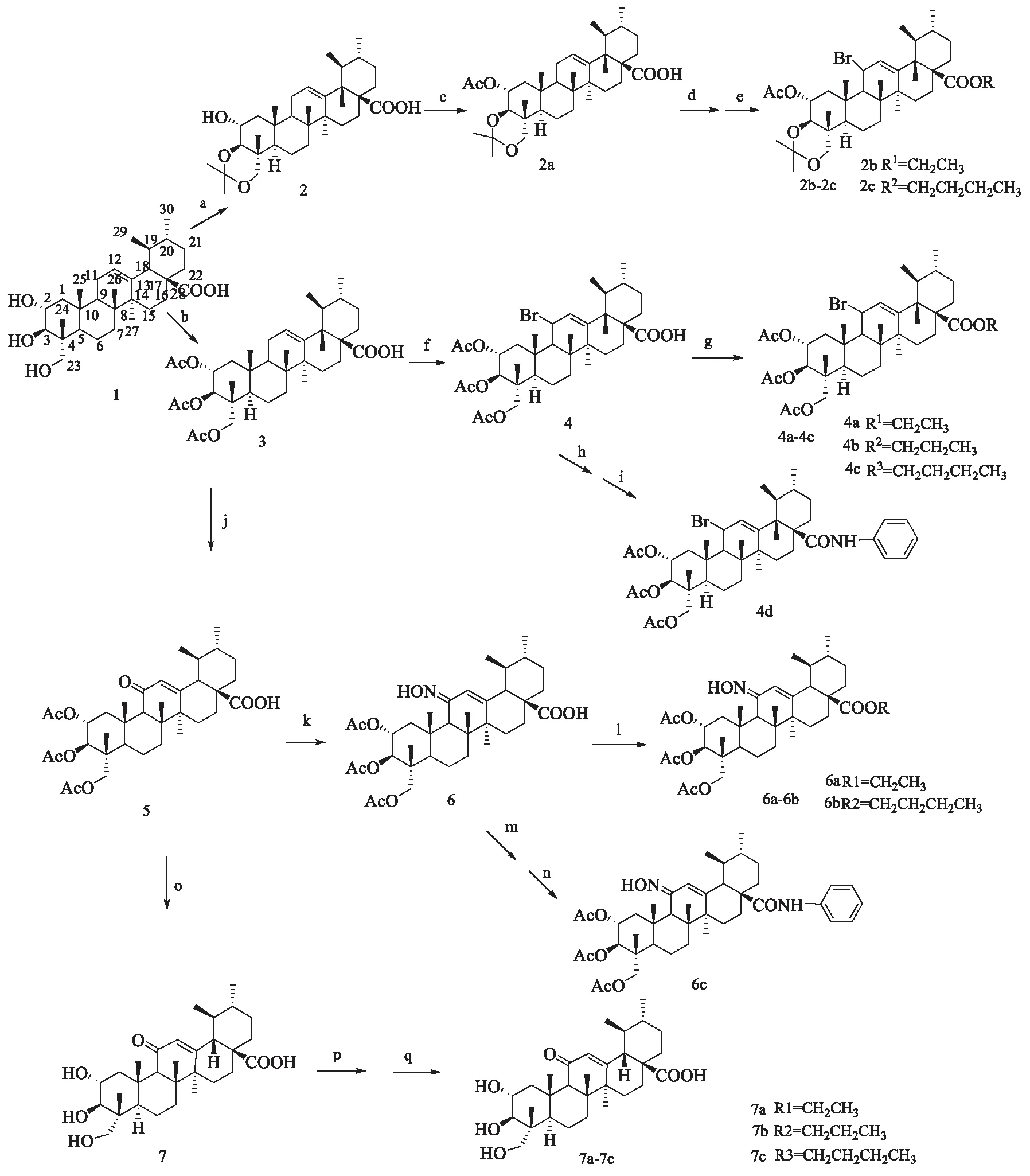
a:(CH3)2C(OCH3)2,P-TsOH,DMF,rt b:Ac2O,DMAP(cat.),THF,rt c:AC2O,DMAP d:K2CO3,NBS,C4H8O2e:K2CO3,BrR,DMF f:K2CO3,NBS,C4H8O2g:K2CO3,BrR,DMF h:CH2Cl2,C2O2Cl2I:aniline,Et3N,CH2Cl2,rt j:K2CrO7/ACOH,reflux k:NHOH.HCl,pyridine,116 ℃,reflux l:BrR,Et3N,CH2Cl2,rt m:(COCl)2,Et3N,CH2Cl2,rt n:aniline,Et3N,CH2Cl2,rt o:NaOH,CH3OH,reflux p:NH2OH.HCl,CH3COONa,C5H5N q:K2CO3,BrR,DMF
图1 目标化合物的合成路线
Fig.1 Synthetic routes of target compounds
1.2.2 2-乙酰氧基-3,23-O-异亚丙基-乌苏烷型-11-溴-12-烯-28-酯类化合物(2b-2c)的合成
由化合物2a经11位溴代后和溴乙烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,得白色粉末状固体(化合物2b)18.2 mg,产率为25.7 %.mp:153.2~156.1 ℃.IR (KBr):2 923 cm-1,2 853 cm-1,1 745 cm-1,1 632 cm-1,1 457 cm-1,1 043 cm-1,602 cm-1.ESI-MS:716.8 (M+H+H2O)+.
由化合物2a经11位溴代后再和溴代正丙烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,得白色粉末状固体(化合物2c)20.2 mg,产率为28.6 %.mp:152.2~156.9 ℃.IR (KBr):2 919 cm-1,2 850 cm-1,1 746 cm-1,1 466 cm-1,1 041 cm-1,601 cm-1.ESI-MS:744.9(M+K)+.
1.2.3 2α,3β,23-三乙酰氧基-乌苏烷型-12-烯-28-羧酸 (3)的合成
在25mL茄形瓶中加入100.0 mg(0.20 mmoL)积雪草酸,加入少量4-二甲氨基吡啶(DMAP),再加入5 mL的THF使其溶解,滴加6 mmoL乙酸酐.室温搅拌.TLC检测反应的终点(显色剂:体积分数95 %的硫酸乙醇溶液,展开剂为V(石油醚)/V(乙酸乙酯)=3/1).反应结束后,蒸干溶剂,充分水洗,过滤,滤饼洗至中性.自然晾干,得产物108 mg,收率为86.80 %.mp:150.8~152.6 ℃.IR(KBr):2 951 cm-1,2 923 cm-1,1 747 cm-1,1 697 cm-1,1 457 cm-1,1 370 cm-1,1 235 cm-1,1 045 cm-1,ESI-MS:632.5(M+H2O)+.
1.2.4 2α,3β,23-三乙酰氧基-乌苏烷型-11-溴-12-烯-28-羧酸 (4)的合成
在25mL茄形瓶中加入50.0 mg(0.08 mmoL)化合物3,加入6 mL的1,4二氧六环使其溶解,搅拌5 min后加入6.64 mg(0.08 mmoL)K2CO3,在光照条件下加入18.0 mg(0.1 mmol)NBS,室温搅拌2 h.母液以15 mL水稀释后,用乙酸乙酯提取(4×4 mL),合并乙酸乙酯层,依次以1 mol/L稀盐酸,饱和NaHCO3,饱和食盐水洗涤,无水Na2SO4干燥,抽滤,减压蒸馏.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,得白色粉末状固体(化合物4)32.3 mg,产率为57.68 %.IR(KBr):2 941 cm-1,2 875 cm-1,1 743 cm-1,16 23 cm-1,1 236 cm-1,1 032 cm-1.mp:140.2~142.1 ℃.ESI-MS:707.5(M+H)+.
1.2.5 N-[2α,3β,23-三乙酰氧基-乌苏烷型-11-溴-12-烯-28-酰]-酯类化合物(4a-4c)的合成
由化合物4和溴乙烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=7/1,得白色粉末状固体(化合物4a)23.7 mg,产率为40.86 %.mp:143.2~144.1 ℃.1H-NMR(300 MHz,CDCl3):δ5.6(m,1H,H-12),δ4.1(m,2H,—COOCH2—),δ3.5~3.9(s,2H,—CH2OH),δ2.3~2.4(m,3H,—CH—),δ1.6~1.9(m,2H,—CH2—),δ2.0~2.1(s,15H,—OAC),δ1.7(m,2H,—CH—),δ1.2~1.5(m,14H,—CH2×7),δ1.3(s,3H,CH3),δ1.25(s,9H,CH3×3),δ0.6~0.9(m,9H,CH3×3).IR(KBr):2 930 cm-1,2 873 cm-1,1 747 cm-1,1 633 cm-1,1 233 cm-1,1 042 cm-1,601 cm-1.ESI-MS:722.3(M+H)+.
由化合物4和溴代正丙烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=7/1,得白色粉末状固体(4b)16.2 mg,产率为27.93 %.mp:145.6~146.9 ℃.IR(KBr):2 975 cm-1,2 851 cm-1,1 741 cm-1,1 632 cm-1,1 460 cm-1,1 039 cm-1,642 cm-1.ESI-MS:734.2(M)+.
由化合物4和溴代异丁烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=7/1,得白色粉末状固体(4c)15.7 mg,产率为27.07 %.mp:148.2~150.1 ℃.1H-NMR(300 MHz,CDCl3):δ5.6(s,1H,H-12),δ4.0(m,2H,—COOCH2—),δ3.6~3.8(s,2H,—CH2OH),δ2.3~2.4(m,3H,—CH—),δ1.5~2.2(m,6H,—CH2CH2CH2—),δ2.0(s,15H,—OAC),δ1.7(m,2H,—CH—),δ1.2~1.4(m,14H,—CH2×7),δ1.3(s,3H,CH3),δ1.25(s,9H,CH3×3),δ0.6~0.9(m,9H,CH3×3).IR(KBr):2 929 cm-1,2 871 cm-1,1 741 cm-1,1 633 cm-1,1458 cm-1,1 034 cm-1,601 cm-1.ESI-MS:747.5(M-H)+.
1.2.6 N-[2α,3β,23-三乙酰氧基-乌苏烷型-11-溴-12-烯-28-酰]-胺类化合物(4d)的合成
在25 mL茄形瓶中加入0.100 g(0.16 mmoL)的2,3,23-三乙酰氧基-11-溴-乌苏烷型-12-烯-28-羧酸,加入3 mL二氯甲烷使其充分溶解,再缓慢滴加0.64 mmoL的草酰氯,室温搅拌24 h.反应结束后,减压蒸干溶剂,加入3 mL环己烷,溶液呈浑浊状态,蒸干溶剂.重复该操作3次,得相应的三乙酰氧基-11-肟基-积雪草酸的酰氯粗品.用3 mL二氯甲烷溶解,滴加三乙胺,调节溶液的pH值为8~9,然后加入苯胺0.05 mL(0.64 mmoL).室温搅拌.TLC检测反应终点.反应结束后,蒸干溶剂,加入适量的水稀释,2 mol/L盐酸调溶液的pH值3~4,充分水洗.过滤,滤饼洗至中性.粗品经柱层析分离纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=4/1,得白色粉末(化合物4d)32.5 mg,收率31.86 %.mp:137.3~139.9 ℃.IR(KBr):2 961 cm-1,1 745 cm-1,1 661 cm-1,922 cm-1,803 cm-1,744 cm-1;ESI-MS:644.1(M+H+H2O)+.
1.2.7 2α,3β,23-三乙酰氧基-乌苏烷型-11-羰基-12-烯-28-羧酸(5)的合成
将50.0 mg (0.08 mmoL) 化合物3与75.0 mg(0.25 mmoL) K2Cr2O7加入到5 mL AcOH中搅拌回流.TLC跟踪检测反应终点.反应结束后,冷却至室温,加入适量H2O稀释,用CH2Cl2萃取2~3次,合并有机相,用饱和NaHCO3溶液洗涤至体系呈弱碱性,分出有机相,再用饱和NaCl溶液洗涤有机相2次,合并有机相,无水MgSO4干燥过夜.抽滤干燥剂,减压蒸除溶剂,得到粗品为淡绿色晶体.粗品经硅胶柱色谱分离纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,减压蒸干洗脱剂得到白色晶体(化合物5)32.1 mg,收率:63.86 %.IR(KBr):2 932 cm-1,1 743 cm-1,1 672 cm-1,1 456 cm-1,1 367 cm-1,1 265 cm-1,1 040 cm-1.mp:288.4~289.6 ℃.ESI-MS:650.3(M+H)+.
1.2.8 2α,3β,23-三乙酰氧基-乌苏烷型-11-肟基-12-烯-28-羧酸 (6)的合成
将250.0 mg (0.08 mmoL) 化合物5溶于适量吡啶中,加入盐酸羟胺6.68 mg(0.096 mmoL).115 ℃下回流1.5 h.TLC跟踪检测反应终点.反应完毕,倒入冰水中,产生大量白色沉淀,抽滤,水洗滤饼,粗品经硅胶柱色谱分离纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,干燥得白色固体(化合物6)26.8 mg,产率为47.54 %.mp:157.3~159.8 ℃;IR(KBr):2 924 cm-1,1 741 cm-1,1 692 cm-1,1 458 cm-1,1 387 cm-1,1 245 cm-1,1 045 cm-1.ESI-MS:644.3(M+H)+.
1.2.9 2,3,23-三乙酰氧基-乌苏烷型-11-肟基-12-烯-28-酯类化合物(6a-6b)的合成
将350 mg(0.075 mmoL)化合物6溶解在4 mL的DMF中,加入少量的碳酸钾,滴入溴乙烷0.023 mL(0.310 mmoL),室温搅拌,用TLC跟踪检测确定反应终点.混合物用乙酸乙酯提取,有机层用蒸馏水及饱和NaCl溶液洗涤,无水MgSO4干燥,抽滤,减压蒸馏得白色固体,粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=9/1,得白色粉末状固体(化合物6a)29.5 mg,产率为58.57 %.mp:170.6~173.2 ℃;IR(KBr):2 931 cm-1,2 873 cm-1,1 745 cm-1,1 660 cm-1,1 455 cm-1,1 369 cm-1,1 237 cm-1,1 040 cm-1.ESI-MS:672.9(M+H)+.
由化合物6和溴代异丁酯反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=7/1,得白色粉末状固体(化合物6b)21.2 mg,产率为40.41 %.mp:178.2~180.1 ℃.1H-NMR(300 MHz,CDCl3):δ5.65(s,1H,H-12),δ4.05(m,2H,—COOCH2—),δ3.6~3.7(m,2H,—CH—),δ3.1(s,2H,—CH2OH),δ2.3~2.4(m,3H,—CH—),δ1.6~1.7(m,6H,—CH2CH2CH2—),δ2.0(s,15H,—OAC),δ1.1~1.8(m,14H,—CH2×7),δ1.3(s,3H,CH3),δ1.2(s,9H,CH3×3),δ0.7~0.9,(m,9H,CH3×3).IR(KBr):2 923 cm-1,2 853 cm-1,1 745 cm-1,1 632 cm-1,1 457 cm-1,1 371 cm-1,1 236 cm-1,1 043 cm-1.ESI-MS:700.3(M+H)+.
1.2.10 2,3,23-三乙酰氧基-乌苏烷型-11-肟基-12-烯-28-酰胺类化合物(6c)的合成
在25 mL茄形瓶中加入0.100 g(0.15 mmoL)化合物6,加入3 mL二氯甲烷使其充分溶解,再缓慢滴加0.64 mmoL草酰氯,室温搅拌24 h.反应结束后,减压蒸干溶剂,加入3 mL环己烷,溶液呈浑浊状态,蒸干溶剂.重复该操作3次,得相应的三乙酰氧基-11-肟基-积雪草酸的酰氯粗品.用3 mL二氯甲烷溶解,滴加三乙胺,调节溶液pH值为8~9,然后加入苯胺0.05 mL(0.64 mmoL).室温搅拌,TLC检测反应终点.反应结束后,蒸干溶剂,加入适量的水稀释,2 mol/L盐酸调溶液pH值为3~4,充分水洗.过滤,滤饼洗至中性.粗品经柱层析分离纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=4/1,得白色粉末(化合物6c)49 mg,收率45.16 %.mp:115.0~118.9 ℃.IR(KBr):2 961 cm-1,1 745 cm-1,1 661 cm-1,922 cm-1,803 cm-1,744 cm-1;ESI-MS (m/z):722.9(M+H)+.
1.2.11 2α,3β,23-羟基-乌苏烷型-11-羰基-12-烯-28-羧酸 (7)的合成
将250.0 mg(0.08 mmoL)化合物5溶于甲醇/四氢呋喃(2 mL/2 mL)溶液中,加入1 mL(0.1 mol/L)氢氧化钠溶液,40 ℃下搅拌,TLC检测反应终点.反应结束,减压蒸出溶剂,加入水分散固体,过滤,滤饼水洗至中性.粗品经柱层析分离纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=2/1,室温干燥,产物为白色粉末状固体(化合物7)30.2 mg,产率75.14 %.mp:142.3~145.6 ℃.IR(KBr):2 924 cm-1,1 741 cm-1,1 692 cm-1,1 655 cm-1,1 458 cm-1,1 245 cm-1,1 046 cm-1.
1.2.12 2α,3β,23-羟基-乌苏烷型-11-肟基-12-烯-28-酯类化合物 (7a-7c)的合成
将150.0 mg(0.08 mmoL) 化合物7溶于适量吡啶中,加入盐酸羟胺6.68 mg(0.096 mmol).115 ℃下回流1.5 h,TLC跟踪检测反应终点.反应完毕,倒入冰水中,产生大量白色沉淀.抽滤,水洗滤饼,干燥得白色固体2α,3β,23-羟基-乌苏烷型-11-肟基-12-烯-28-羧酸20.4 mg,产率为49.28 %.mp:146.3~147.8 ℃;IR(KBr):2 924 cm-1,1 741 cm-1,1 692 cm-1,1 458 cm-1,1 387 cm-1,1 245 cm-1,1 045 cm-1.由化合物2α,3β,23-羟基-乌苏烷型-11-肟基-12-烯-28-羧酸和溴乙烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=7/1,得白色粉末状固体(化合物7a)19.2 mg,产率为44.31 %.mp:151.2~154.1 ℃.IR(KBr):2 920 cm-1,2 851 cm-1,1 725 cm-1,1 660 cm-1,1 463 cm-1,1 265 cm-1.ESI-MS(m/z):585.2(M+K+H)+.
由化合物2α,3β,23-羟基-乌苏烷型-11-肟基-12-烯-28-羧酸和溴代正丙烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=7/1,得白色粉末状固体(化合物7b)18.5 mg,产率为41.34 %.mp:153.2~154.1 ℃.IR(KBr):2 919 cm-1,2 850 cm-1,1 727 cm-1,1 657 cm-1,1 632 cm-1,1 458 cm-1,1 241 cm-1.ESI-MS(m/z):601.5(M+H)+.
由化合物2α,3β,23-羟基-乌苏烷型-11-肟基-12-烯-28-羧酸和溴代正戊烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=7/1,得白色粉末状固体(化合物7c)16.2 mg,产率为34.46 %.mp:133.2~140.1 ℃.1H-NMR(300 MHz,CDCl3):δ5.63(s,1H,H-12),δ4.05(m,2H,—COOCH2—),δ3.4~3.6(m,2H,—CH—),δ3.2(s,3H,—OH),δ2.9(s,2H,—CH2OH),δ2.3~2.4(m,3H,—CH—),δ1.5~2.2(m,6H,—CH2CH2CH2—),δ2.1(m,1H,—NOH),δ1.1~1.8(m,14H,—CH2×7),δ1.3(s,3H,CH3),δ1.25(s,9H,CH3×3),δ0.8~0.9(m,9H,CH3×3).IR(KBr):2 919 cm-1,2 850 cm-1,1 275 cm-1,1 661 cm-1,1 463 cm-1,1 270 cm-1,1 047 cm-1.ESI-MS(m/z):587.9(M)+.
1.3 初步体外细胞毒活性测试
采用MTT法,选用人胃腺癌细胞株SGC-7901,人非小细胞肺癌细胞株A549和人成纤维肉瘤细胞株HT-1080为靶细胞,以吉非替尼和阿霉素为阳性对照物,进行积雪草酸类似物体外细胞毒活性药理评价.
1.3.1 MTT法检测体外抗肿瘤活性原理
MTT法是化学物质噻唑蓝(MTT),能被活细胞线粒体中的琥珀酸脱氢酶还原,生成难溶性蓝紫色结晶物甲瓒(Formazan),沉积在细胞当中,而死细胞无此功能.二甲基亚砜(DMSO)能溶解细胞的蓝紫色沉积物,用酶联免疫检测仪在490 nm波长处测定其吸光度值,可间接反应活细胞数量.在一定细胞数范围内,MTT结晶数形成量与活细胞数线性相关.
1.3.2 实验操作步骤
1.3.2.1 埋板
将生长至对数生长期的细胞消化、离心,制成细胞悬液,吹匀并将3种细胞SGC-7901、A549、HT-1080密度分别调至2×104个/mL、2.5×104个/mL、1×104个/mL,以180 μL/孔接种到96孔板中,接种过程中不断吹匀细胞,保证细胞密度一致,铺板过程要迅速,保证每孔的细胞数目大致相同,将96孔板置于37 ℃、含摩尔分数5 %的CO2、饱和湿度的培养箱中孵育24 h.
1.3.2.2 加药
将药物配制成以完全培养基为溶剂,初始质量浓度为1 μg/L的溶液,将上述溶液稀释成5个剂量组,药物质量浓度分别为500 mg·L-1,50 mg·L-1,5 mg·L-1,0.5 mg·L-1,0.05 mg·L-1.取各个剂量的药液加入96孔板中,每孔20 μL,每剂量3个复孔.加药后的药物终质量浓度为50 mg·L-1,5 mg·L-1,0.5 mg·L-1,0.05 mg·L-1,0.005 mg·L-1.加药完毕放入培养箱中孵育72 h.
1.3.2.3 MTT检测
将加药并于培养箱中孵育72 h后的96孔板中培养基轻轻甩出,然后加入配置好的MTT溶液(5 g·L-1),每孔20 μL,放入培养箱中继续孵育3 h.取出孵育3 h的96孔板,将其中的MTT溶液轻轻甩出,每孔加100 μL的DMSO,微量振荡仪上振荡5 min以使甲瓒完全溶解,用酶标仪在490 nm下测光密度(OD)值,按下列公式计算药物对肿瘤细胞体外增殖的抑制率(Inhibition Rate,IR):

2 结果与讨论
2.1 化学合成
以积雪草酸为先导化合物,对其2位、3位、12位、23位和28位进行结构修饰,设计并合成12个积雪草酸类似物.目标产物在反应过程中采用TLC监测反应终点,柱层析分离后,其结构通过IR,MS,1H-NMR得以确证.
2.2 生物活性评价
采用四甲基偶氮唑盐法(MTT比色法),选用人胃腺癌细胞株SGC-7901,人非小细胞肺癌细胞株A549和人成纤维肉瘤细胞株HT-1080为靶细胞,以吉非替尼和阿霉素为阳性对照物,进行积雪草酸类似物体外细胞毒活性药理评价.实验结果见表1.细胞株图见图2、图3.
由表1数据可知:积雪草酸衍生物对3种肿瘤细胞有一定抑制作用.化合物4b,6a,6b对SGC-7901、A549、HT-1080的抑制作用均高于AA,其中化合物6b在质量浓度为50 mg·L-1时对SGC-7901、A549、HT-1080的抑制率分别为98.9 %,95.4 %,99.9 %,明显优于对照品吉非替尼(93.1 %,56.5 %,98.2 %)与ADM的抑制活性基本相同.而化合物6a在质量浓度为50 mg·L-1时对于SGC-7901、A549、HT-1080的抑制率分别为33.8 %,30.0 %,58.5 %,具有较弱的抑制活性.化合物4b在质量浓度为50 mg/L时对SGC-7901、A549、HT-1080的抑制率分别为90.1 %,52.4 %,98.6 %,与对照品吉非替尼(93.1 %,56.5 %,98.2 %)抑制活性相当,抑制活性一般.化合物6a(IC50:4.323 μmol·L-1,2.63 μmol·L-1,1.077 5 μmol·L-1)对3种细胞系SGC-7901、A549、HT-1080的细胞毒性与吉非替尼(3.217 μmol·L-1,15.869 μmol·L-1,1.511 5 μmol·L-1)相当,其中对A549的细胞毒性明显优于吉非替尼,对HT-1080的细胞活性略优于吉非替尼.
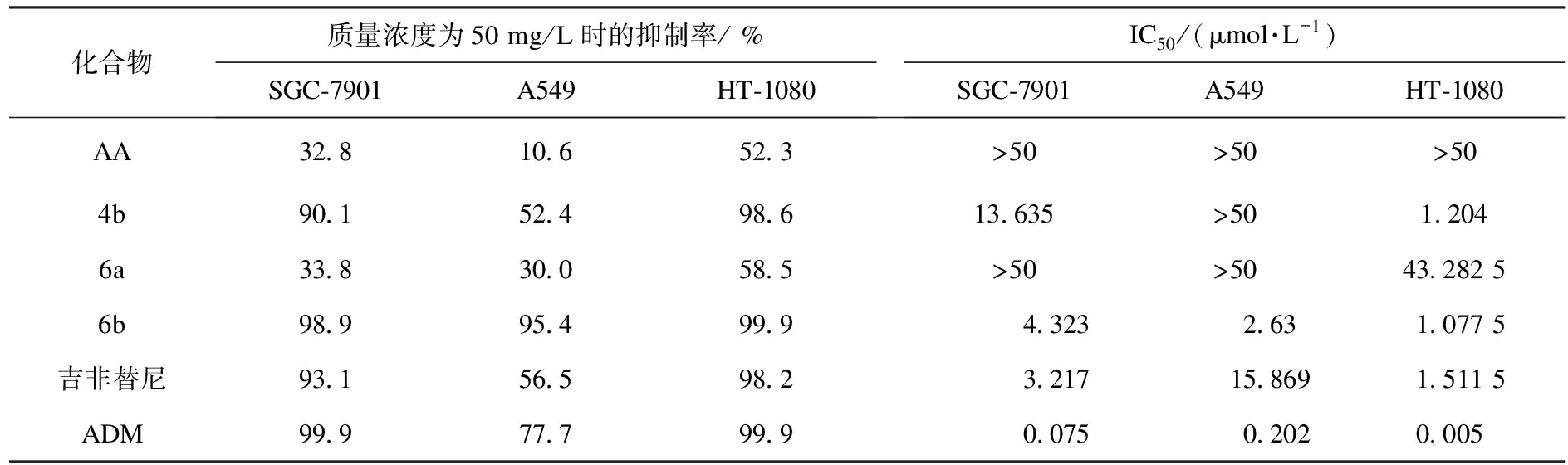
表1 积雪草酸衍生物对SGC-7901、A549、HT-1080的抑制活性

图2 未经药物处理的A549细胞株

图3 用不同浓度的4b处理A549 细胞株48 h的外观形态
3 结 论
初步的药理实验结果说明,积雪草酸12位成肟之后抗肿瘤活性提高,但是被溴取代后抗肿瘤活性降低.28位酯基对积雪草酸的抗肿瘤活性也有一定的影响.研究结果对积雪草酸的结构进一步修饰具有一定的指导意义.
参考文献:
[1] 周军,高静,方春钱.积雪草酸对肝癌细胞增殖作用的影响[J].江苏大学学报:医学版,2009,19(6):475-479.
[2] Huang Y H,Zhang S H,Zhen R X,et al.Asiaticoside Inducing Apoptosis of Tumor Cells and Enhancing Anti-tumor Acitivty of Vincristine[J].Chin J Cancer,2004,23(12):1599-1604.
[3] 汤丽霞,杨光,谭家驹.积雪草酸抑制HSC2-T6细胞Ⅰ型胶原蛋白表达[J].第四军医大学学报,2007,28(13):1178-1180.
[4] 熊御云.积雪草酸对帕金森样的损伤细胞和突变果蝇的神经保护作用[D].江苏:江苏大学,2009.
[5] 李铁军,陆波,邱彦,等.积雪草酸及其衍生物在制备抗抑郁药物中的应用:中国,ZL200310108854.3[P].2004-11-10.
[6] Wen X,Zhang P,Liu J,et al.Pentacyclic Triterpenes.Part 2:Synthesis and Biological Evaluation of Maslinic Acid Derivatives as Glycogen Phosphorylase Inhibitors[J].Bioorg.Med.Chem.Lett.,2006,16(3):722-726.
[7] 范怡梅,高静,徐力致,等.2α,23-二羟基乌苏酸用于制备抗炎药物的用途:中国,CN1543963[P].2004-11-10.
