环丙酮分子及离子的解离的研究
2014-03-20崔金玉
崔金玉
(绥化学院电气工程学院,绥化152061;吉林大学原子与分子物理研究所,长春130012)
1 引 言
酮类也是抗菌药物的主要成分,如第三代喹诺酮类药物常用的有氧氟沙星、环丙沙星等曾被广泛使用,1988 年以来研究出第四代喹诺酮类药物,如莫西沙星、格帕沙星、加替沙星等抗菌作用更强.研究环丙酮的性质,有利于提炼新的药物成分.环丙酮是极不稳定的化合物,国内外关于环丙酮的研究报道不是很多,早在1966 年,Hammond[1]和DeBoer等[2]用乙烯酮和重氮甲烷在氟三氯甲烷中成功制备了环丙酮.2007年艾月洁等人[3]结合实验运用量子化学计算方法CASSCF,B3LYP 和MP2研究在292~365nm 波长的光的激发下,从键长键角的角度研究环丙酮被激发至激发态S1,然后αC-C键断裂.并且找到在αCC 键断裂途径上,存在基态和第一激发势能面的交叉点TS1,然后发生另一个C-C 键的断裂,生成基态产物一氧化碳和乙烯.我们用密度泛函理论B3LYP和TS方法计算环丙酮的解离过程[4,5].我们的工作一是把分子的解离过程更清晰的描述出来,另外还对离子的解离过程做了计算.主要从计算过渡态角度,用IRC 方法查看过渡态连接的反应物和生成物,确定反应的方向.对环丙酮分子稳定的基态计算,找到两个过渡态,一个过渡态是C-C单键断裂,欲成开环结构,再由这样的开环结构,计算得另一C-C键断裂,形成CO 和C2H4(这一过程相当于把环丙酮分子看成等腰三角形,两腰断裂).另一过渡态是环丙酮的另一个C-C键(相当于等腰三角形的底边)断裂,形成异构体,由异构体解离成C2H2O 和CH2.对环丙酮离子构型的计算,离子构型是不稳定构型,也是一种过渡态,找到其开环结构的稳定离子构型.
2 基础理论和计算方法
量子力学理论是适用于微观领域的基本理论,微观领域的一切现象都可以用量子力学的知识来解释.所以,我们利用现代量子化学的方法和原子分子理论来研究分子问题[6].用密度泛函理论[7]UB3LYP方法解决这一问题.密度泛函理论是计算较大分子体系结构的强有力的计算工具,它比Hartree Fock(简称HF)和Moller Plesset(简称MP)方法节省时间.同样的方法计算频率值.在过渡态transition state(简称TS)附近开始用opt=TS优化,初始的TS我是利用固定反应坐标进行优化得到的,用freq 进行TS 判据.同时,IRC计算确认过渡态是反应物和产物之间的过渡态.利用计算机软件Gaussian03,采用基组6-31+G(d,p)首先opt优化环丙酮分子的几何构型,得到稳定的几何构型,计算频率,无虚频,是稳定构型[10-12].结构见图1中.
3 计算结果
3.1 对分子的计算
环丙酮分子式为C3H4O,用Gaussion03软件计算其几何构型,结构见图2 中所示,能量-191.8966Ha,无虚频.其振动频率及相对强度如表1所示.从表1中可见频率为1930.79cm-1时,其振动幅值最大,达358.371KM/Mole.其次是频率964.151cm-1,强度为108.244 KM/Mole.另外,在频率为1018.02cm-1时,强度为27KM/Mole.在频率为1072.33cm-1时,强度为15KM/Mole.其它情况很弱,不去分析,仅对这几个频率作分析如下:

表1 环丙酮分子的基态频率和相对强度Table 1 Frequency and Intensity of Cyclopropanone molecule
(1)在频率freq=1930.79cm-1时,强度最大358.371KM/Mole.此时1号O 原子和2号C 原子振动最强烈,可能它们与分子其它部分断开,形成CO 和C2H4,也可能O 与C之间断开,形成O 和C3H4.但是,这两种情况都没有找到过渡态[8],可能O 与C之间双键结构很难断开,另外,2C-3C和2C-4C双键同时断开也很难,也没有找到双键同时断开的过渡态,所以很可能先断开单键,成开环结构,然后再断开另一个C-O 键,见下面(2)的分析情况.
(2)在频率freq=964.15cm-1时,强度为108.242KM/Mole,振动幅度也比较大,分析其振动情况[9],2C左右摆动幅度很大,可能会使分子的2C-4C或2C-3C之间的键断裂,成开环结构.因为分子对称,两种断裂情况的几率相同,断裂后的分子结构、振动情况都相同,所以只分析一种情况即2C-4C 键断裂.寻找这样的过渡态[13],得到TS1,见图1 中所示,其能量为-191.8350 Ha,有一个虚频值为-642.68cm-1.分析此时的振动情况[14],2C向外振动剧烈,很可能2C-3C之间键断裂,并且用IRC 优化TS2,所得两个方向的结果,一个方向可以生成反应物,把另一个方向的结果继续优化,得产物CO 和C2H4的共同体,不是稳定构型,能量为-191.9171 Ha,进一步分解成产物CO 和C2H4.这一过程见图1中所示.

图1 环丙酮分子的解离过程(Ⅰ)Fig.1 Dissociation of Cyclopropanone molecule(Ⅰ)
(3)在频率为-1018.02cm-1时,强度为27 KM/Mole,也进行了分析,3C-4C 各向分子外振动,寻找3C-4C键断裂的过渡态TS2,结构见图2中所示,虚频率为-525.1544cm-1,能量为-191.8287Ha,IRC计算表示,它是反应物和异构体INT1的中间过渡态,异构体INT1见图2中,是稳定构型,能量为-191.8799 Ha.由INT1的振动模式分析,2C与3C各向分子两侧振动剧烈,欲使2C与3C之间键断开,找到这种情况的过渡态TS3,图2中所示,能量为-191.6539 Ha,虚频值为-46.53cm-1,由此生成产物C2H2O 和CH2共同体,无虚频是稳定构型[15].但这一TS3势垒较高,欲解离成产物C2H2O 和CH2远不如(2)的情况容易.

图2 环丙酮分子的解离过程(Ⅱ)Fig.2 Dissociation of cyclopropanone molecule(Ⅱ)
(4)频率为1072.33cm-1时的情况,强度为15KM/Mole.3C-4C 振动方向仍相反,但与(3)情况不同,不是向分子外振动,而是上下振动,分子欲发生扭转,可能2C-3C 或2C-4C 之间断裂,情况如2情况相似;也可能3C-4C 键断裂[16],情况同(3)情况相似.
3.2 对离子的异构化分析计算
将环丙酮分子的稳定结构的简正坐标作为输入,对离子结构进行优化[4,5,17],最后得到过渡态(TS4),能量为-191.5612 Ha,有一个虚频为-285.23cm-1,说明环丙酮离子极不稳定,观察虚频下的振动模式,O 和2C一起向离子左右两边振动,且振动强烈,可能2C-3C键断开,也可能2C-4C键断开,两种情况相同,只分析一种,得离子的开环结构,即稳定的离子的异构体INT2(2C-4C键断开).见图3中所示.能量为-191.5798 Ha,无虚频稳定.分析INT2振动最强烈的情况,1O和2C向离子外振动强烈,计算过渡态,得到过渡态TS5,频率为-164.15 cm-1,能量值为-191.5525Ha.IRC 显示TS5是INT2和INT3(2C-3C键断开)之间的过渡态.最后得离子的稳定构型就是开环结构INT2和INT3.

图3 环丙酮离子的异构化Fig.3 Isomerism of Cyclopropanone ion
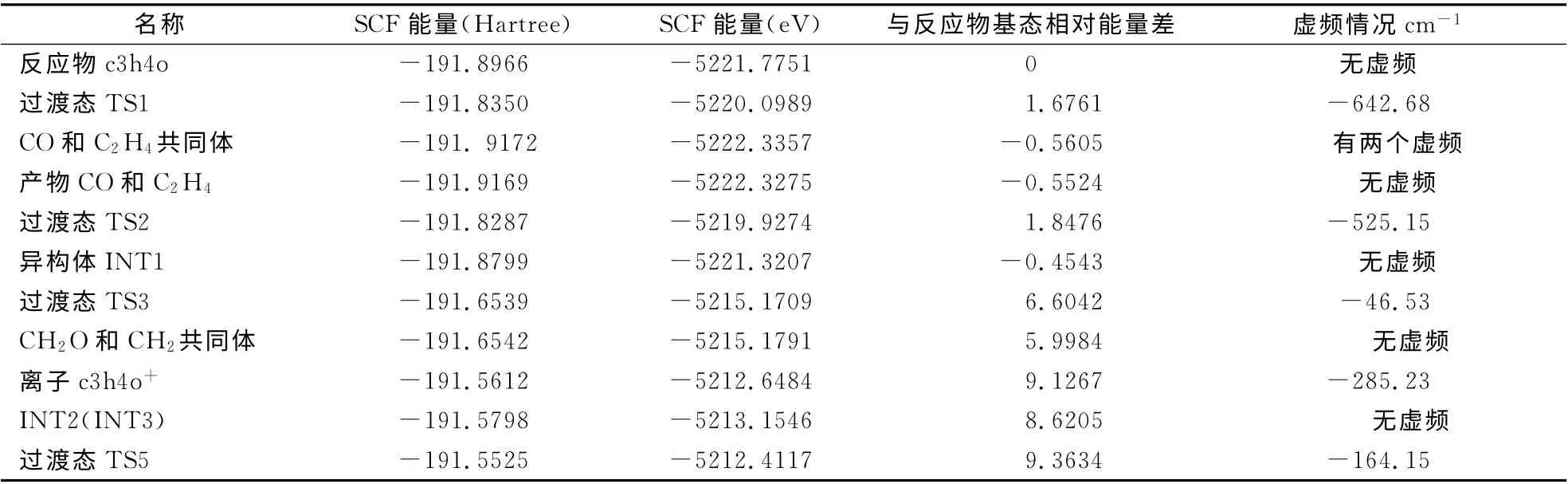
表2 环丙酮分子到解离产物及其异构体的各状态能量值(Hartree)及频率(cm-1)情况(1Ha=27.2114eV) Table 2 Energy(Hartree)and frequency(cm-1)of from Cyclopropanone molecule to product and isomer(1Ha=27.2114eV)
4 结 语

图4 环丙酮分子解离过程的势能面Fig.4 The energies surface of Dissociation of Cyclopropanone molecule and Ion
最后得出环丙酮分子及离子解离过程各状态能量高低见图4所示,能量大小见表2中.
本文用密度泛函DFT(B3LYP)计算环丙酮分子及离子稳定结构,并用TS计算过渡态.从计算结果看出,环丙酮分子的解离产物CO 和C2H2,这与艾月洁等人[3]计算结果相同.但是,计算结果表明还有产物C2H2O 和CH2.但从能量上看,前者容易实现(TS1=-191.8350eV),后者过渡态能量大(TS3=-191.6539eV),所以实现起来有些困难,但在能量足够大的时候,会有产物C2H2O 和CH2生成.另外,实现前者时,单键容易断开,双键同时断开很难,没有找到双键同时断开的过渡态,但找到单键断开的过渡态及解离到产物的过程.同样的方法计算环丙酮离子C3H4O+,它是不稳构型,有一个虚频值,而它的开环异构体稳定,我们找到它的异构化过程.所以从以上计算表明,环丙酮离子异构体稳定,分子解离产物是一氧化碳和乙烯,及存在异构体,在能量足够高时,解离成亚甲基CH2和C2H2O 共同体.
[1] Turro N J,Hammond W B.Tetramethylcyclopropanone,Ⅱ.Mechanism of the Favorskii Rearrangment[J].J.Am.Chem.Soc.,1965,87:3258.
[2] Schaafsma S E,Steinberg H,Deboer T J.The synthesis of cyclopropanone[J].Rec.Trav.Chim.,1966,85,1170.
[3] Ai Y J,Lin L,Fang W H.Ab inito study mechanisms of photodissociation and thermal isomerization of cyclopropanone[J].Acta Chim.Sin.,2007,65(2):129(in Chinese)[艾玥洁,林 玲,方维.环丙酮光解离和热异构机理的从头算研究[J].化学学报,2007,65(2):129]
[4] Cui J Y,Jin MX,Wang Z G,et al.Dissociation of 2-methyl-cyclopentanone Ion[J].At.Mol.Phys.,2009,26(5):787(in Chinese)[崔金 玉,金明星,王志刚,等.2-甲基环戊酮离子的解离[J].原子与分子物理学报,2009,26(5):787]
[5] Cui J Y.Dissociation of the first six excited states of 2-methyl-cyclopentanone ion excited state[J].J.Mol.Sci.,2011,27(3):199(in Chinese)[崔金玉.2-甲基环戊酮离子前六个激发态的解离[J].分子科学学报,2011,27(3):199]
[6] Tang A Q,Yang Z Z,Li Q S.Quantum chemistry[M].Beijing:Science Press,1982.(in Chinese)[唐敖庆,杨忠志,李前树.量子化学[M].北京:科学出版社,1982]
[7] Jiang F L.Principles of quantum chemistry [M].Shanghai:Fudan University Press,1990(in Chi-nese)[江逢林.量子化学原理[M].上海:复旦大学出版社,1990]
[8] Roothan C C J,Bagus P S.Methods of molecular quantum mechanics[M].Academic Press,1964.
[9] Gunnarsson O,Jones R O.Density Functional Calculations for Atoms,Molecules and Clusters[J],Phys.Scr.,1980,21:394.
[10] Becke A D.Density-functional thermochemistry.Ⅲ.the role of exact exchange[J].Chem.Phys.,1993,98:56.
[11] Lee C,Yang W,Parr R G.Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density[J].Phys.Rev.B,1988,37:785.
[12] Simons J P.The photochemical and spectroscopic approach to molecular reaction dynamics[J].Comments on At.Mol.Phys.,1985,16(3):157.
[13] Leone S R.Laser probing of chemical reaction dynamics[J].Science,1985,227.
[14] Bernstein R B.Chemical dynamics via molecular beam and laser techniques[M].New York:Oxford University Press,1982.
[15] Smith I V M.Kinetics and dynamics of elementary gas reaction[M].London:Butter Worth,1980.
[16] Wang Z G,Pan S F,Yao M G.The two hydrogen transfer dissociation channels of nicotine [J].J.Mol.Struct.:Theochem.,2006,767:99.
[17] Jin M X,Cui Q L,Emad Mukhtar,et al.Secondharmonic-generation measurements on ZnSe under high pressure [J].J.Phys.:Condens.Matt.,2002,14:11037.
