NO和HSO偶联及其异构化反应机理
2014-02-28王智香
王智香,曹 佳
(延安大学化学与化工学院,陕西延安716000)
NO和HSO偶联及其异构化反应机理
王智香,曹 佳*
(延安大学化学与化工学院,陕西延安716000)
采用G2M(CC5)//MPW1PW91/6-311+G(2df,p)方法,研究了HSO和NO偶联及其异构化反应机理,获得了各物种的几何构型和频率数据,并构建了标题反应的势能剖面。结果表明,该反应存在3条不同路径,优势路径为R(NO+HSO)→IM1→TS1→IM2,其表观活化能为42.38 kJ· mol-1。此过程为NO中N原子与HSO中O原子偶合形成中间体IM1,接着IM1中SH基团从O(1)原子迁移到O(2)原子上后反应完成。
HSO;NO;偶联;异构化机理
在煤深加工和石油炼制过程产生的挥发性有机物对大气环境具有严重危害[1-3]。研究这些挥发物大气反应机理,对降解和消除此类污染物具有重要的实际意义[4]。HSO是大气中一种挥发小分子。目前已对HSO和其它自由基或小分子(如H,O,OH,O2,O3,NO2和NO等)的大气反应做了广泛研究[5,6]。NO也是大气重要组分,平流层含量较多[7]。实验结果认为HSO和NO反应的速率常数小于1.0×10-15cm3·molecule-1·s-1,但并未研究其内在机理[6]。更为重要的是,对于HSO和NO自由基均含未成对电子,二者偶联反应是大气中重要反应过程。遗憾的是,尚未见HSO和NO的结合及其产物进一步异构化的机理研究报道。本文对HSO和NO的偶联和产物异构化机理做细致研究,为实验研究提供重要理论参考。
1 计算方法
应用MPW1PW91/6-311+G(2df,p)方法对所有物种进行构型优化和频率计算。根据内禀反应坐标(IRC)验证过渡态是否连接到正确的反应物和产物。此外,用G2M(CC5)方法对各个物种做了高水平单点能校正,构建了势能面[8]。上述计算由Gaussian 03程序计算完成[9]。
2 结果与讨论
2.1 方法准确性验证
图1给出了MPW1PW91/6-311+G(2df,p)方法计算得到的所有物种几何构型。由于仅有HSO和NO有实验构型参数,将理论结果与相应文献值比较发现,计算的NO中N-O键长为0.1140 nm,实验值为0.1154 nm,偏差为0.0014 nm[10]。对于HSO,计算的键角∠HSO为104.2°,实验值为106.6°,二者相差2.4°;计算的HSO中H-S和SO键长分别为0.1374和0.1500 nm,相应实验值分别为0.1389和0.1494 nm,相差分别为0.0015和0.0006 nm[11]。此外,表1列出了计算的所有物种的频率数据。由表1数据可得,所有中间体频率均为实频,为势能面稳定点。过渡态有且仅有一个虚频,为势能面鞍点[12]。由上结果可以看出,本文应用MPW1PW91/6-311+G(2df,p)方法计算的频率和构型数据是正确的。

图1 用MPW1PW91/6-311+G(2df,p)方法优化所得的所有物种的几何构型[键长:nm,键角:(°)]
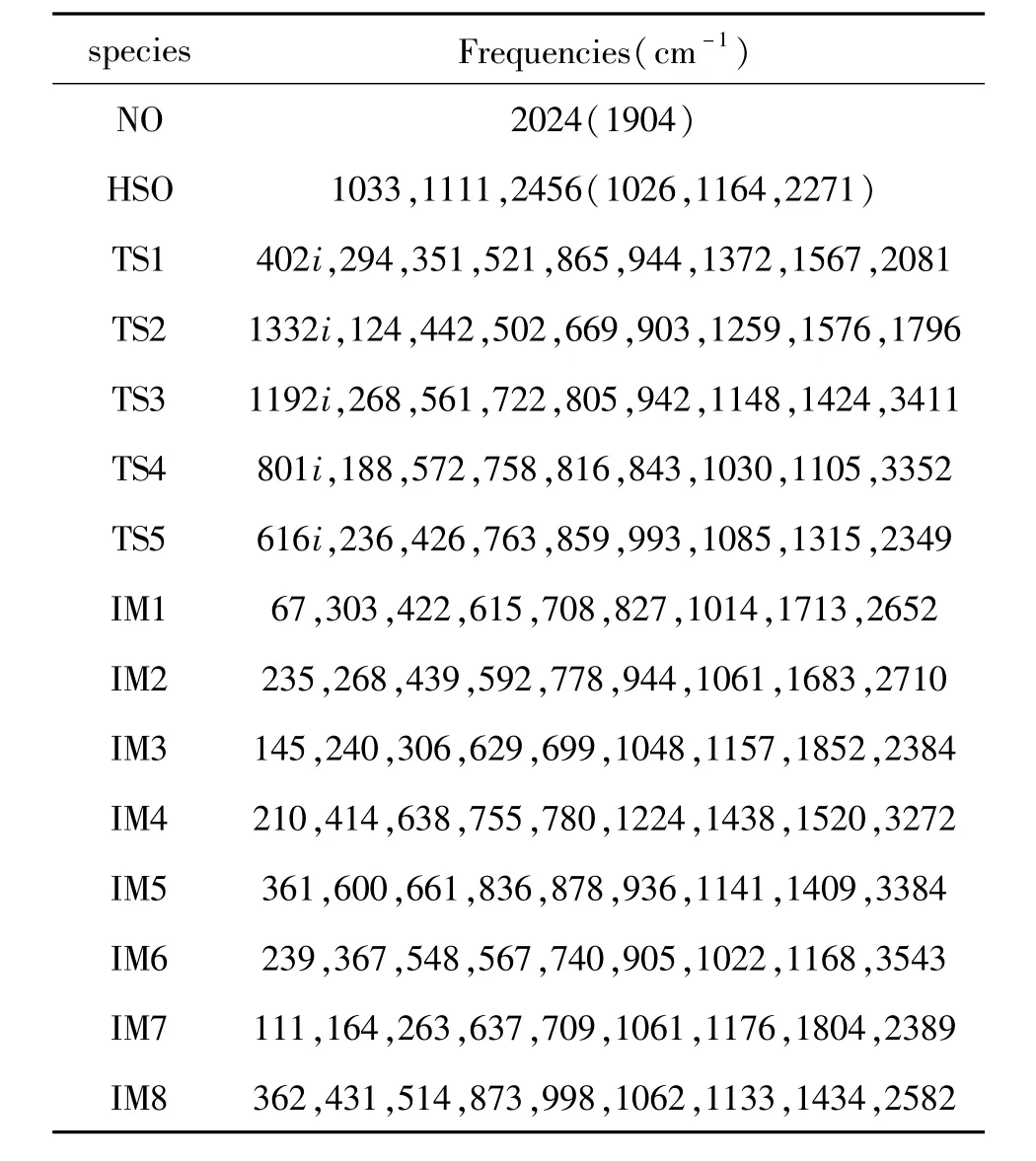
表1 MPW1PW91/6-311+G(2df,p)方法计算的所有物种的振动频率

图2 HSO和NO反应方案
2.2 反应物静电势分析
根据图3中HSO和NO的静电势,对二者可能的偶合方式进行了探索。图3中红色区域表明大的负电势,而蓝色区域意味着正电势出现。HSO中O和H原子周围分别环绕强的负电势和正电势,S原子则处于正负电势之间。在NO中,N和O原子均处于负电势和正电势之间。根据静电势作用原理,正负电势之间彼此容易偶合,同时又受到原子电负性和分子空间结构等多种因素影响。故本文计算获得了以NO中N原子进攻HSO中S以及O原子的优势方式为出发点,进一步对其偶合及异构化反应机理做详细研究。
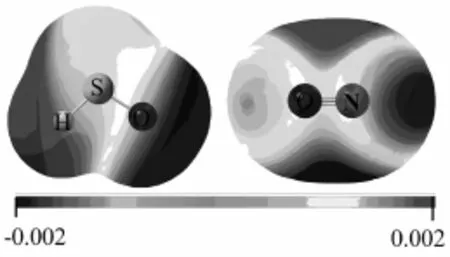
图3 HSO和NO的静电势图
2.3 中间体异构化机理
2.3.1 NO的N原子进攻HSO的O原子
如图1和图4所示,NO中N原子进攻HSO中O(1)原子,当O(1)-N键缩短至0.1379 nm,S-O(1)键拉伸至0.1696 nm,键角∠NO(1)S为120.7°时,形成中间体IM1,释放热量37.45 kJ·mol-1。接着需克服表观活化能为42.38 kJ·mol-1到达过渡态TS1。在TS1中,S原子靠近O(2)原子远离O(1)原子,键角∠NO(1)S缩小至100.1°,S-O(1)键长继续拉伸至0.2094 nm,而S-O(2)键长变为0.2317 nm。振动频率分析发现S-H基团在O(1)和O(2)原子之间振动。最后,S-H基团由O(1)原子迁移到O(2)上形成中间体IM2。IM2与IM1互为顺反异构,二者构型参数非常接近,只是H原子的空间方向不同,IM1中H原子趋向于环内侧,而IM2中H原子在过渡态环外侧。IM1和IM2相对于反应物(HSO+NO)的能量分别为-37.45和-36.18 kJ·mol-1。总的来说,该过程无论是直接偶合还是后续异构化,由于表观活化能较低,容易发生。
2.3.2 NO的N原子顺式进攻HSO的S原子
路径[R(HSO+NO)→IM3→TS2→IM4→TS3→IM5→TS4→IM6]从NO中的N原子进攻HSO中的S原子出发,当HSO中N和S原子间距离为0.2044 nm,∠O(2)NS为106.0°时,形成中间体IM3。在IM3中O(1)和O(2)原子分布在以N-S键为轴的同一个方向,故称为N和S原子的顺势进攻。紧接着H原子从S原子迁移到N原子发生,形成过渡态TS2。此时,断裂的H-N键为0.1322 nm,形成的H-S键长0.1631 nm。H原子迁移完成后形成中间体IM4。能量角度,中间体IM3和IM4的相对反应物能量分别为-32.42和-93.99 kJ·mol-1,过渡态TS2表观活化能为77.13。由此看出,IM3容易形成,IM4为热力学方面容易形成,由于TS2高的表观活化能,为动力学控制步骤。
在IM4形成之后,反应还可以实现两步异构化过程,分别由IM4中O(1)与O(2)原子强的相互做用过渡态TS3形成趋向于环状的不稳定中间体IM5,以及IM5中O(2)原子进一步由N原子迁移到O(1)原子,最后形成中间体IM6。虽然此过程也是可能的异构化过程,但由于中间体大的相对能(IM5,259.51 kJ·mol-1;IM6,150.65kJ·mol-1),而过渡态(TS3,299.36 kJ·mol-1;TS4,347.68 kJ· mol-1)具有高的表观活化能,异构化很难实现,故这里不作详细分析。

图4 反应HSO和NO在G2M(CC5)//MPW1PW91/6-311+G(2df,p)水平上的势能面图
2.3.3 NO的N原子反式进攻HSO的S原子
此路径首先形成中间体IM7,放出能量15.77 kJ·mol-1。IM7是由NO中N原子进攻HSO中S原子后,O(1)原子与O(2)原子以N-S键为轴呈反式方向。当S-N键长为0.1584 nm,键角∠O(2)NS为77.7°时,到了过渡态TS5。随后O(2)原子从N原子迁移至S原子上,形成了中间体IM8。该中间体结构中∠O(2)-S-N为69.4°,∠O(2)SO(1)为124.9°,O(2)-S键长为0.1517 nm,N-S键长为0.1543 nm。反应本质是O(2)原子从N原子迁移至S原子上,O(2)-N键的断裂,O(2)-S键的生成。该过程由于TS5的高表观活化能为225.33 kJ·mol-1,很难实现IM8形成,只是停留在中间体IM7形成。
综上所述,上述3条路径中,由路径[R(HSO+NO)→IM3→TS2→IM4]容易发生,为主反应路径。
3 结论
本文采用G2M(CC5)//MPW1PW91/6-311+G(2df,p)方法对NO中N原子分别进攻HSO中S与O的偶合反应以及后续异构化过程机理作了详细研究,找到了8个中间体和5过渡态。结果表明NO中N原子与HSO中O结合能力强于和O原子结合。
致谢:感谢陕西师范大学理论与计算化学研究室提供的结果分析讨论和机群资源支持。
[1]周东,何玉凤,黄志新.化工区域挥发性有机物污染控制研究[J].广州化工,2012,40(14):157-159.
[2]刘新宇,张萍,修光利,等.上海市石油化学和化工行业环境污染新问题及监控对策[J].化学世界,2013(9):565-569.
[3]姚磊,马嫣,陈敏东.亚甲基环己烷气相臭氧氧化反应[J].环境化学,2013,32(10):1834-1840.
[4]赵燕,王慧,张孝敏,等.OH抽提1-戊醇分子中α-H和β-H引发的大气反应机理的理论研究[J].化学学报,2009,67(2):122-128.
[5]Atkinson R,Baulch D L,Cox R A,et al.Evaluated kinetic and photochemical data for atmospheric chemistry:Volume I-gas phase reactions of Ox,HOx,NOx and SOx species[J].Atmos.Chem.Phys,2004(4):1461-1738.
[6]Selim H,Gupta A K,Sassi M.Novel error propagation approach for reducing H2S/O2 reaction mechanism[J].Applied Energy,2012,93:116-124.
[7]李晓燕,刘群,郑世钧.HOSO+NO反应的机理及动力学[J].物理化学学报,2011,27(3):564-570.
[8]Mebel Am,Morokuma K,Lin M C.Modification of the GAUSSIAN-2 theoretical model:The use of coupledcluster energies,density-functional geometries,and frequencies[J].J.Chem.Phys,1995,103(17):7414-7421.
[9]Frisch M J,et al.Gaussian 03,Revision C.02,Gaussian,Inc,Wallingford CT.2004.
[10]Russelld,Johnson III.NIST Computational Chemistry Comparison and Benchmark Database and NIST Standard Reference Database Number 101 Release16a.August 2013.
[11]Wang N X,Wilson A K.Density Functional Theory and the Correlation Consistent Basis Sets:The Tight d Effect on HSO and HOS.J.Phys.Chem.A 2005,109:7187-7196.
[12]卢彦霞,王文亮,王渭娜,等.CH3SO+HO2气相反应机理与主通道速率常数[J].化学学报,2010,68(13):1253-1260.
[责任编辑 李晓霞]
Coupling and Its Isomeric M echanism of HSO and NO
WANG Zhi-xiang,CAO JIA*
(School of Chemistry&Chemical Engineering,Yanan University,Yanan 716000,China)
Coupling and isomeric mechanism of NO and HSO are calculated at the G2M(CC5)//MPW1PW91/6-311+G(2df,p)level.Geometries and frequencies of all the species are obtained,and potential energy surface is constructed.The result shows that the reaction has three paths,and themajor path is R→IM1→TS1→IM2 with the apparent activation energy of 42.38 kJ·mol-1.The detailed process of the dominant path is the coupling of N atom in NO with O atom in HSO,leading to the formation of IM1.Subsequently,the transfer of SH group in IM1 from O(1)atom to O(2)atom.
HSO;NO;coupling;isomeric mechanism
O641
A
1004-602X(2014)03-0045-04
10.13876/J.cnki.ydnse.2014.03.045
2014-07-05
陕西省教育厅专项科研计划项目(2013JK0667);延安大学青年项目资助(YDQ2013-16);延安大学化学与化工学院自然科学专项基金资助(YDHG2014-12)
王智香(1983—),女,河北沧州人,延安大学助理实验师。 *为通讯作者
