多孔金属有机骨架材料储氢性能分子模拟
2013-09-21吴选军蔡卫权
吴选军 郑 佶 李 江 蔡卫权
(武汉理工大学化学工程学院,武汉430070)
1 引 言
化石能源面临枯竭,可再生替代能源逐渐成为未来能源战略重点,氢能源因来源广泛和清洁性而成为重要替代能源之一.1−3为提高储氢密度和安全性,各种储氢材料应运而生,如金属氢化物、4,5笼状结构水合物、6,7金属有机骨架结构(MOFs)材料、8−10共价有机骨架结构(COFs)材料11,12和多孔芳香骨架结构(PAFs)材料13,14等均被用于储氢性能的测试,其中具有多孔特性和超高比表面积的MOFs材料最具应用潜力.15,16MOFs是通过金属或金属氧化物与有机配体自组装而形成的一种具备规整结构的多孔材料,改变金属或金属氧化物骨架和有机联接配体能够形成一系列具有不同拓扑结构和不同物理化学性质的多孔骨架材料,在气体储存、分离与催化和分子传感等领域具有广阔的应用前景.8−10
近十年来,MOFs材料的储氢性能研究受到广泛关注,Yaghi研究组8−10首先在实验室合成了一系列具有高比表面积的规整结构MOFs材料,通过N2吸附实验表明某些MOFs材料具有非常高的比表面积,如MOF-177和MOF-210的比表面积分别高达4500和6240 m2·g−1,远远超过活性炭和分子筛等传统高比表面积吸附材料.17这些具有超高比表面积的多孔骨架材料表现出良好的储氢性能,如IRMOF-1在78 K下的储氢量达到4.5%(w),MOF-177在77 K和7.0 MPa下的储氢量更高达7.5%(w).16,17
但研究发现,MOFs材料的储氢量并不十分严格地随着其比表面积的增大而增加,其自由孔体积以及吸附剂分子与H2分子之间的相互作用等因素对其储氢能力也有显著影响.18,19为了研究各种因素对MOFs材料储氢性能的影响,Snurr研究组19−24对一系列MOFs材料的储氢性能进行巨正则系综蒙特卡洛(GCMC)模拟,确定如果要使MOFs材料在298 K和12 MPa下满足美国能源部2010年的储氢标准(6%(w)),对自由孔体积在1.6−2.4 cm3·g−1的多孔结构材料而言,其等量吸附热(反映H2与吸附剂之间的相互作用力强弱的指标)必须达到10−15 kJ·mol−1,但目前所有MOFs材料对H2分子的等量吸附热远低于此标准.19
为了提高MOFs材料与H2分子间的相互作用从而增大储氢量,Yaghi研究组25最近合成了由较短有机配体或可以形成互穿结构的瘦长有机配体与金属氧化物骨架自组装形成的具有较小孔径的新型MOFs材料,该类材料具有较高的储氢能力,如MOF-324在77 K下的储氢量可达4.9%(w).25为了理解此类材料的储氢机理,通过GCMC模拟从分子层面分析其与H2分子之间的相互作用具有重要的理论意义,但迄今尚无文献报道此类材料的力场参数.
Frost和Snurr等18−24研究表明,DREIDING 力场能够很好地描述一部分MOFs材料的气体吸附性能,但同一套参数并不适合所有的MOFs材料.26−28本文在DREIDING力场基础上,通过参数优化,提出适合于具有互穿结构的MOFs材料的力场参数,通过GCMC分子模拟验证力场参数在H2吸附方面的准确性,并对IRMOF-1、IRMOF61和IRMOF62等几种不同结构的MOFs材料的储氢机理进行比较与分析.
2 模型与计算
2.1 模型与力场参数
IRMOF-1、IRMOF61和IRMOF62的晶胞结构见图1.它们均由Zn4O骨架与有机酸联结体组成,其中联结体分别为对苯二甲酸、乙炔基二苯二甲酸和丁二炔基二苯二甲酸,其中后两种MOFs材料具有互穿结构.所有MOFs材料初始构型均来源于剑桥晶体数据库,29具体结构参数见表1.
2.1.1 力场参数
采用GCMC分子模拟研究MOFs材料的储氢性能时,可将MOFs材料骨架原子固定不动,通常不会对吸附结果造成影响.18−24因此,GCMC分子模拟时仅需考虑MOFs材料与吸附质分子间相互作用和吸附质与吸附质分子之间的相互作用,而这两种相互作用常采用如下伦纳德-琼斯(Lennard-Jones,LJ)位能函数描述:18

式(1)中的相同原子LJ参数取至Frost等18优化的DREIDING力场参数,不同原子之间的LJ相互作用EVDW参数εij和σij采用Lorentz-Bertelot混合规则(式2)计算,18结果见表2.

2.1.2 GCMC模拟

图1 IRMOF-1、IRMOF61和IRMOF62的金属骨架、有机联结体及晶体结构Fig.1 Metal frameworks,organic linkers,and crystal structures of IRMOF-1,IRMOF61,and IRMOF62

表1 IRMOF-1、IRMOF61和IRMOF62材料的结构参数Table 1 Structural parameters of IRMOF-1,IRMOF61 and IRMOF62
采用GCMC方法模拟H2分子在MOFs材料中的吸附特性,模拟程序为Snurr等28提供的MUSIC-4.0程序代码.为了节省计算资源,除IRMOF-1模拟盒子包含2×2×2超级晶胞外,IRMOF-61模拟盒子包含2×2×1超级晶胞,而IRMOF-62模拟盒子只包含单元晶胞,经实际测试,这种处理方法对吸附结果没有影响.骨架结构采用刚性模型,H2分子则采用Buch全原子模型,30其LJ位能参数见表2.LJ相互作用截断半径为1.28 nm,各个方向均采用周期性边界.模拟总循环步数为107步,前5×106步为平衡时间,后5×106步为采样时间.

表2 MOFs材料的力场参数Table 2 Force field parameters of MOF materials
3 结果与讨论
3.1 力场参数的可靠性
3.1.1 IRMOF-1的储氢性能
为了检验力场参数及晶体模型的可靠性,采用GCMC方法对不同温度下IRMOF-1材料的H2分子等温吸附曲线进行模拟,模拟的力场LJ参数按Frost等18报道的文献数据(见表2)计算.由于H2分子采用全原子模型,所以计算时不用考虑静电相互作用.
图2为77 K、不同压力下H2在IRMOF-1材料中的等温总吸附曲线和等温过量吸附曲线.图2(a)所用数据为绝对吸附量(标注为abs.,下同),即GCMC模拟达到热力学平衡时的总吸附量值;而图2(b)所用数据则为过量吸附量值(标注为exc.,下同).两者可通过式(3)进行转换:18

式(3)中Nexc.和Nabs.分别为过量吸附量与绝对吸附量,ρbulk和Vp分别为吸附质分子在主体相中的密度和吸附骨架的自由体积.这里吸附质H2的密度采用Peng-Robinson状态方程计算,31而吸附骨架的自由体积按Myers and Monson报道的方法计算,32即:

从图2中曲线可以看出,本文模拟得到的77 K、不同压力下IRMOF-1材料对H2的等温吸附曲线均与文献9,18报道的实验或模拟数据相符,表明本文计算方法可靠.
3.1.2 IRMOF-61和IRMOF-62的储氢性能

图2 77 K、中高压条件下H2在IRMOF-1材料中的等温绝对(abs.)和过量(exc.)总吸附曲线Fig.2 Absolute(abs.)or excess(exc.)adsorption isotherms of H2in IRMOF-1 at 77 K
图3为77 K、中高压条件下H2分子在IRMOF-62中的等温过量吸附曲线(以下简称等温吸附曲线).从图3可以看出,如果直接采用Frost等18报道的力场LJ参数(适用于IRMOF-1等多孔骨架材料的H2分子吸附)来进行GCMC模拟,达到热力学平衡后的过量摩尔吸附量比Tranchemontagne等25报道的实验数据超出约20%,出现明显的过估.
为了让GCMC模拟结果与实验结果相符,本文先调低了H2分子间的LJ能量参数取ε/kB=34.2 K,LJ原子直径参数不变;并借鉴了Pérez-Pellitero等26提出的方法,即在原力场LJ参数的基础上乘以适当的校正系数,可以改善UFF力场对沸石咪唑酯骨架材料(ZIFs)的气体吸附量出现过估的情况.本文选择以下校正公式:

式(5)保留了原来DREIDING力场中的LJ原子直径参数,将其LJ能量参数适当降低.利用校正后的力场LJ能量参数进行GCMC模拟后发现,预测的H2吸附等温线与实验数据十分吻合,证明校正后的力场LJ参数是可行的.由于在低压下H2分子吸附量的变化更能体现H2分子与多孔骨架之间的相互作用,而在高压下H2分子吸附量受多孔骨架材料的比表面积或自由孔体积影响更大,18本文又通过GCMC模拟了低压下H2分子在IRMOF-62中的吸附性能.

图377 K、中高压条件下H2在IRMOF-62中的等温吸附曲线Fig.3 Excess adsorption isotherms of H2in IRMOF-62 at 77 K and medium and high pressures

图4 77 K、低压条件下H2在IRMOF-62中的等温吸附曲线Fig.4 Excess adsorption isotherms of H2in IRMOF-62 at 77 K and low pressure
图4为77 K、低压条件下H2在IRMOF-62中的等温吸附曲线.与Tranchemontagne等25报道的实验数据相比,本文模拟的等温吸附数据是可靠的.
为了进一步验证本文力场参数的可移植性,又对77 K、不同压力下H2在IRMOF-61中的等温吸附曲线进行预测(见图5).

图6 298 K下H2在不同MOFs材料中的等温吸附曲线Fig.6 Excess adsorption isotherms of H2in different MOF materials at 298 K

图5 77 K下H2在IRMOF-61中的等温吸附曲线Fig.5 Excess adsorption isotherms of H2in IRMOF-61 at 77 K
从图5中曲线可以看出,77 K、不同压力下H2在IRMOF-61材料中的等温吸附曲线变化趋势与IRMOF-62基本一致.但与文献25报道实验数据相比,77 K、低压下H2在IRMOF-61材料中的等温吸附曲线模拟结果存在较大偏差,只有在100 kPa压力条件下的H2吸附量与实验数据相符.采用本文力场参数对相同条件下模拟得到的H2分别在IRMOF-61材料中的等温吸附曲线与在IRMOF-62材料中的等温吸附曲线变化趋势相似,而文献25报道两者的等温吸附曲线实验数据变化趋势相差较大,造成这种差异的原因还有待进一步确证.由于迄今为止尚无77 K、中高压下H2在IRMOF-61材料中的等温吸附曲线实验数据的文献报道,因此无法对相应结果的预测质量进行判断.
3.1.3 常温储氢性能
由于燃料电池的储存和释放气体H2的条件为−40−85°C和0.15−10.0 MPa,11因此研究在此温度和压力范围内多孔材料的储氢性能具有现实意义.本文对三种不同MOFs材料在298 K温度下对H2的等温吸附曲线进行了GCMC模拟,结果见图6.
从图6中曲线可以看出,IRMOF-61和IRMOF-62在298 K下的储氢能力并没有提高,低于IRMOF-1在同等条件下的储氢能力.由于迄今为止还没有IRMOF-61和IRMOF-62在298 K下储氢量实验数据的报道,因此本文预测的数据还有待进一步实验验证.
3.2 储氢位置
3.2.1 低压下H2的吸附位置
Frost等18的研究表明,对相同拓扑结构的MOFs骨架材料而言,H2在低压区的吸附量主要依赖于等量吸附热Qst,在中等压力区的吸附量依赖于MOFs材料的比表面积,而在高压区的吸附量则依赖于自由孔体积.本文计算了不同MOFs材料的比表面积和自由孔体积,比表面积采用Düren等21提出的方法计算,而自由孔体积按式(4)计算,结果见表1.不同MOFs材料对H2的吸附热Qst按式(6)计算:18

式(6)中R和T分别表示气体常数与吸附温度;U为吸附相的势能;N为已吸附的吸附质分子数; 表示巨正则系综平均值.77和298 K、10 kPa下H2在不同MOFs材料中的等量吸附热的计算结果见表3.
表3显示,77 K、10 kPa下H2在IRMOF-1与IRMOF-62材料中的等量吸附热与文献18,25数据相符,等量吸附热与等温吸附量变化规律也相同,其中,IRMOF-62具有最高的等量吸附热与等温吸附量,IRMOF-1与IRMOF-61的数据大致相当;但H2在IRMOF-61中的等量吸附热偏低,这与前面预测低压下H2在IRMOF-61中的等温吸附量偏低的结果相一致,说明本文力场对IRMOF-61的储氢能力存在低估,这可能与该力场没有单独考虑炔烃基与H2的相互作用有关,下一步工作可对此进行优化.298 K和10 kPa下H2在IRMOF-61与IRMOF-62材料中的等量吸附热尚无文献报道,但本文计算结果与等温吸附曲线变化规律一致.

表3 10 kPa下H2在MOFs材料中的等量吸附热Table 3 Isosteric adsorption heat of H2in various MOFs at 10 kPa

图7 77 K、10 kPa下H2在MOFs中的吸附平衡构象Fig.7 Conformation snapshots of H2adsorption equilibrium in MOFs at 77 K and 10 kPa
为进一步理解MOFs材料与H2之间的相互作用机理,本文利用VMD软件33制作了77 K、10 kPa下H2在不同MOFs材料中达到吸附平衡时最后一帧构象抓图(见图7).图7(a)的构象与文献18一致,H2会优先吸附在Zn4O骨架附近靠近苯环的位置;图7(b)和图7(c)的构象显示,H2也会优先吸附在Zn4O骨架附近靠近苯环的位置.
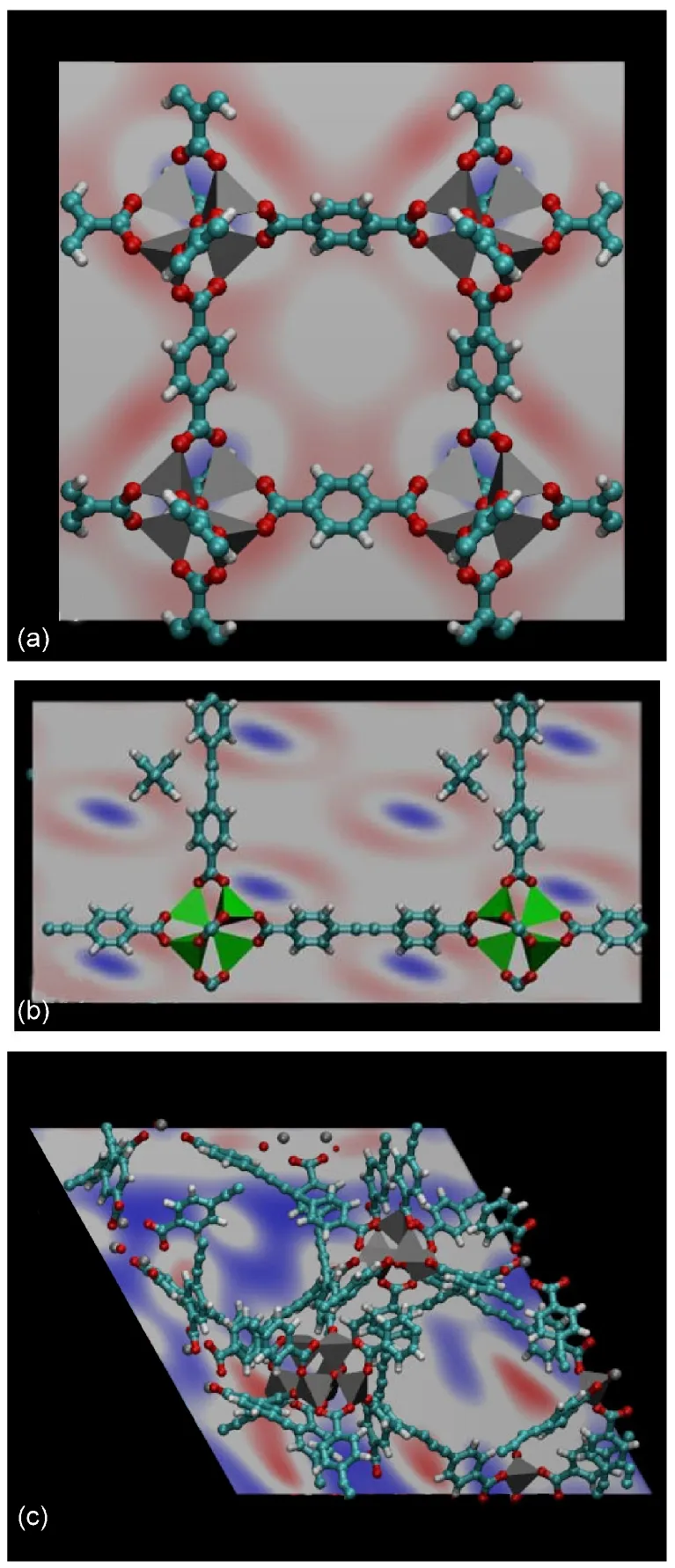
图8 77 K、3.0 MPa下H2在MOFs材料中的吸附几率密度分布图Fig.8 Probability density distribution for adsorption of H2in MOFs at 77 K and 3.0 MPa
3.2.2 中高压下H2的吸附位置
为了得到中高压下H2在MOFs材料中吸附平衡时的几率密度分布数据,本文采用自编程序对GCMC结果进行统计计算.将MOFs单元晶胞至少划分为50×50×50个同样大小的格子,然后统计每个吸附分子在格子中心出现的几率大小,即为标准高斯密度分布,并逐个累加起来.为了保证满足所有格子中的累积几率为1的条件,最后将各个格子中累加的几率数据进行了归一化处理.由于IRMOF-62属于三斜晶体,为了让格子中心坐标范围与单元晶胞一致,采用了Düren等21提出的坐标转换方法进行处理.由吸附平衡数据处理好后的几率密度分布数据按VMD固定格式导入,采用VMD软件制作出最后的几率密度分布图.34

图9 77 K下H2在MOFs材料中的等温绝对吸附量曲线Fig.9 Absolute adsorption isotherms of H2in MOFs at 77 K
图8 为77 K、3.0 MPa下H2在不同MOFs材料中处于吸附平衡时的几率密度分布图,图示为不同二维平面(见图注)切片,切片偏移量相对值为0.5.图8中蓝色区域为H2吸附的低密度区,红色区域为H2吸附的高密度区,而白色区域为H2吸附的中等密度区.从图8可以看出,压力3.0 MPa下H2在IRMOF-1材料中的优先吸附位置与低压下的情况一致,即H2优先吸附在Zn4O骨架附近靠近苯环的位置,随着压力的增加,H2在IRMOF-1材料中的吸附量逐渐增大,H2分子吸附位置向孔腔中心偏移,但依然存在非常明显的优先吸附区域(图中呈现红色“8”字型),表明孔腔中心存在空心的吸附死角.另外,图中蓝色区域为苯环占据的空间,蓝色区域的取向代表苯环的偏转方向,从图中所示的蓝色区域的取向和苯环的偏转方向可以看出,相邻网格苯环的偏转方向相互垂直.
对具有互穿结构的MOFs材料而言,由于其孔腔尺寸缩小,使得H2分子优先吸附位区域零散化,IRMOF-61中H2分子优先吸附区域呈现椭圆形,不同椭圆形区域之间不存在交叉重叠,归因于瘦长型的有机配体加大了Zn4O骨架之间的距离,代表吸附低密度区的蓝色区域取向平行.IRMOF-62中H2分子优先吸附区域更加零散化,代表吸附低密度区的蓝色区域明显扩展而互相连接在一起,说明在IRMOF-62中存在很多H2分子吸附死角.IRMOF-62中H2分子优先吸附区域依然集中在Zn4O骨架附近靠近苯环的位置,而丁二炔基团对H2的吸附较弱,在其附近形成了很多H2吸附死角.
实际应用时多孔材料对H2的绝对吸附量更能反映出其真实的储氢能力.35为了研究不同MOFs材料对H2吸附能力的影响,本文比较了77 K下H2在IRMOF-1、IRMOF-61和IRMOF-62材料中的等温绝对吸附量曲线(见图9).由于在IRMOF-62中存在很多吸附死角,与其它两种MOFs材料相比,其H2绝对吸附量明显偏小.而IRMOF-61中乙炔基较短,对H2分子吸附没有造成太大影响,反而因为其较小的孔腔尺寸增大了其对H2的吸附能力.文献25报道IRMOF-61和IRMOF-62在77 K温度下对N2分子的等温吸附曲线也表现出类似的规律.
4 结论
对传统DREIDING力场参数进行了优化,该力场能够在全压力范围内很好地复制H2分子在IRMOF-62材料中的等温吸附曲线,而采用优化前的力场参数将出现20%左右的过估;但该力场对H2分子在IRMOF-61中的等温吸附曲线预测出现低估.同时对常温下H2在不同MOFs材料中的等温吸附曲线进行了预测,与IRMOF-1相比,两种具有互穿骨架结构的MOFs材料IRMOF-61和IRMOF-62的常温储氢能力并无明显提高.通过比较低压和中高压条件下H2在MOFs材料中达到吸附平衡时的几率密度分布图发现,H2会优先吸附在Zn4O骨架附近靠近苯环的位置,对具有互穿结构的MOFs材料而言,由于其孔腔尺寸缩小,使得H2分子优先吸附位区域零散化;由于有机配体尺寸过长,导致在IRMOF-62中产生很多吸附死角,从而弱化了IRMOF-62对H2的吸附能力,而适当长度的有机配体能保证既形成互穿骨架结构并增强与H2分子间的相互作用,又能避免形成过多吸附死角,因此具备较高的储氢能力.
致谢: 特别感谢Snurr教授提供MUSIC-4.0程序代码进行GCMC模拟,同时感谢可视化工具软件VMD等开源软件的作者Humphrey教授等,本文工作完成离不开上述软件的支持.
(1) Rowsell,J.L.C.;Yaghi,O.M.J.Am.Chem.Soc.2006,128,1304.doi:10.1021/ja056639q
(2)Wong-Foy,A.G.;Matzger,A.J.;Yagh,O.M.J.Am.Chem.Soc.2006,128,3494.doi:10.1021/ja058213h
(3) Nijem,N.;Veyan,J.F.;Kong,L.Z.;Li,K.H.;Pramanik,S.;Zhao,Y.G.;Li,J.;Langreth,D.;Chabal,Y.J.J.Am.Chem.Soc.2010,132,1654.doi:10.1021/ja908817n
(4) Yang,J.;Sudik,A.;Wolverton,C.J.Phys.Chem.C 2007,111,19134.doi:10.1021/jp076434z
(5) Skipper,C.V.J.;Hoang,T.K.A.;Antonelli,D.M.;Kaltsoyannis,N.Chem.-Eur.J.2012,18,1750.doi:10.1002/chem.v18.6
(6) Lu,H.L.;Wang,J.W.;Liu,C.L.;Ratcliffe,C.I.;Becker,U.;Kumar,R.;Ripmeester,J.J.Am.Chem.Soc.2012,134,9160.doi:10.1021/ja303222u
(7) Senadheera,L.;Conradi,M.S.J.Phys.Chem.B 2007,111,12097.doi:10.1021/jp074517+
(8) Eddaoudi,M.;Kim,J.;Rosi,N.;Vodak,D.;Wachter,J.;O'Keeffe,M.;Yaghi,O.M.Science 2002,295,469.doi:10.1126/science.1067208
(9) Rowsell,J.L.C.;Spencer,E.C.;Eckert,J.;Howard,J.A.K.;Yaghi,O.M.Science 2005,309,1350.doi:10.1126/science.1113247
(10) Li,H.;Eddaoudi,M.;O'Keeffe,M.;Yaghi,O.M.Nature 1999,402,276.doi:10.1038/46248
(11) Han,S.S.;Furukawa,H.;Yaghi,O.M.;Goddard,W.A.J.Am.Chem.Soc.2008,130,11580.doi:10.1021/ja803247y
(12) Lan,J.H.;Cao,D.P.;Wang,W.C.J.Phys.Chem.C 2010,114,3108.doi:10.1021/jp9106525
(13) Sun,Y.X.;Ben,T.;Wang,L.;Qiu,S.L.;Sun,H.J.Phys.Chem.Lett.2010,1,2753.doi:10.1021/jz100894u
(14) Ben,T.;Ren,H.;Ma,S.;Cao,D.;Lan,J.;Jing,X.;Wang,W.;Xu,J.;Deng,F.;Simmons,J.M.;Qiu,S.;Zhu,G.T.Angew.Chem.Int.Edit.2009,48,9457.doi:10.1002/anie.200904637
(15) Chae,H.K.;Siberio-Pérez,D.Y.;Kim,J.;Go,Y.;Eddaoudi,M.;Matzger,A.J.;O'Keeffe,M.;Yaghi,O.M.Nature 2004,427,523.
(16) Rosi,N.L.;Eckert,J.;Eddaoudi,M.;Vodak,D.T.;Kim,J.;O'Keefee,M.;Yaghi,O.M.Science 2003,300,1127.doi:10.1126/science.1083440
(17) Furukawa,H.;Ko,N.;Go,Y.B.;Aratani,N.;Choi,S.B.;Choi,E.;Yazaydin,A.Ö.;Snurr,R.Q.;O'Keeffe,M.;Kim,J.;Yaghi,O.M.Science 2010,329,424.doi:10.1126/science.1192160
(18) Frost,H.;Düren,T.;Snurr,R.Q.J.Phys.Chem.B 2006,110,9565.doi:10.1021/jp060433+
(19) Frost,H.;Snurr,R.Q.J.Phys.Chem.C 2007,111,18794.doi:10.1021/jp076657p
(20) Dalach,P.;Frost,H.;Snurr,R.Q.;Ellis,D.E.J.Phys.Chem.C 2008,112,9278.doi:10.1021/j9801008d
(21) Düren,T.;Millange,F.;Ferey,G.;Walton,K.S.;Snurr,R.Q.J.Phys.Chem.C 2007,111,15350.doi:10.1021/jp074723h
(22) Bae,Y.S.;Snurr,R.Q.Microporous Mesoporous Mat.2010,132,300.doi:10.1016/j.micromeso.2010.02.023
(23) Bae,Y.S.;Snurr,R.Q.Microporous Mesoporous Mat.2010,135,178.doi:10.1016/j.micromeso.2010.07.007
(24) Getman,R.B.;Miller,J.H.;Wang,K.;Snurr,R.Q.J.Phys.Chem.C 2011,115,2066.doi:10.1021/jp1094068
(25) Tranchemontagne,D.J.;Park,K.S.;Furukawa,H.;Eckert,J.;Knobler,C.B.;Yaghi,O.M.J.Phys.Chem.C 2012,116,13143.doi:10.1021/jp302356q
(26) Pérez-Pellitero,J.;Amrouche,H.;Siperstein,F.R.;Pirngruber,G.;Nieto-Draghi,C.;Chaplais,G.;Simon-Masseron,A.;Bazer-Bachi,D.;Peralta,D.;Bats,N.Chem.-Eur.J.2010,16,1560.doi:10.1002/chem.v16:5
(27) Pantatosaki,E.;Pazzona,F.G.;Megariotis,G.;Papadopoulos,G.K.J.Phys.Chem.B 2010,114,2493.doi:10.1021/jp911477a
(28) Gupta,A.;Chempath,S.;Sanborn,M.J.;Clark,L.A.;Snurr,R.Q.Mol.Simul.2003,29,29.doi:10.1080/0892702031000065719
(29) The Cambridge Crystallographic Data Centre.http://www.ccdc.cam.ac.uk(accessed March 2013).
(30) Buch,V.J.Chem.Phys.1994,100,7610.doi:10.1063/1.466854
(31) Peng,D.Y.;Robinson,D.B.Ind.Eng.Chem.Fund.1976,15,59.doi:10.1021/i160057a011
(32) Myers,A.L.;Monson,P.A.Langmuir 2002,18,10261.doi:10.1021/la026399h
(33) Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graph.1996,14,33.doi:10.1016/0263-7855(96)00018-5
(34)Wu,X.J.;Yang,X.;Song,J.;Cai,W.Q.Acta Chim.Sin.2012,70,2518.[吴选军,杨 旭,宋 杰,蔡卫权.化学学报,2012,70,2518.]doi:10.6023/A12110858
(35) Han,S.S.;Choi,S.H.;Goddard,W.A.J.Phys.Chem.C 2011,115,3507.doi:10.1021/jp200321y
