杯[4]吡咯-卤素、铵离子复合物的理论研究
2013-07-25陈雪松路鹏飞董玉慧
陈雪松 路鹏飞 董玉慧 解 菊,*
(1扬州大学化学化工学院,江苏扬州 225002; 2潍坊科技学院,山东潍坊 262700)
1 引言
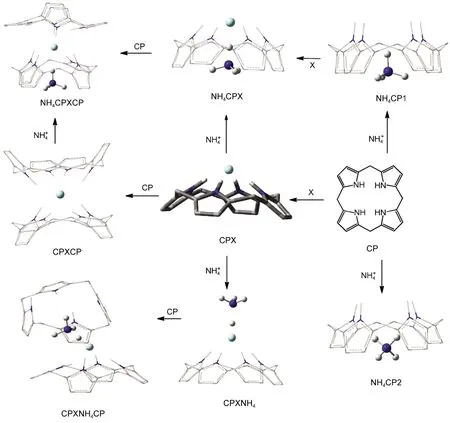
图1 杯[4]吡咯(CP)与卤素阴离子(X-=F-,Cl-,Br-)和铵根离子可能的组装体系Fig.1 Possible complexes between calix[4]pyrrole(CP)and halide anions(X-=F-,Cl-,Br-)and ammonium ion
杯吡咯大环化合物作为一类易于制备且识别性能优异的阴离子1及中性小分子2受体而备受人们关注.在阴离子键合、传感器、3色谱分离技术以及药物传输4等方面的研究显示了其潜在的研究价值和应用前景.杯吡咯是由吡咯环和sp3杂化碳原子通过吡咯环的α位连接而组成的一类非共轭结构的柔性大环化合物.目前,合成的杯吡咯大环化合物基本上都是以杯[4]吡咯为母体环,即环体中含有4个吡咯单元.杯[4]吡咯存在四种构象,无论是在自由状态、固相还是极性溶剂中,均是1,3-交替构象能量最低.5当存在与之相互作用的其它底物分子或离子(如卤素阴离子)时,无论是液相或是固相,环体则采取锥式构象.杯[4]吡咯头部具有N―H结构,易于阴离子形成很好的N―H…X氢键作用,是一种理想的阴离子受体;其空腔的柔性结构,可实现对不同构型的阳离子进行包合,从而形成结构新奇的杯[4]吡咯超分子组装体系.
计算机模拟研究在杯吡咯化学领域起到了揭示实验现象、预测实验结果、指导实践应用的重要作用.尤其是对于杯[4]吡咯的构象、6与阴离子及离子对的键合作用、7-10与阴离子作用的动态过程5,11等方面的研究,更是成为杯吡咯化学研究领域的热点课题.杯吡咯与阴离子以及离子对的主-客体相互作用仍然属于弱相互作用,因而需要精度较高的计算模拟方法.在该领域的相关文献中应用最广泛的B3LYP方法在描述氢键以及范德华力等方面却存在不足之处.12本文致力于杯[4]吡咯与离子形成的超分子体系的系统研究.采用先进的M06-2X13密度泛函方法结合精细的图形显示工具,考察各种可能的组装模式(见图1),为杯吡咯化学的研究与应用提供一定的理论依据.
2 计算方法
杯吡咯与阴离子之间最主要的相互作用是氢键,因此计算方法的选择尤为重要.密度泛函理论方法经过几十年的发展,在分子间弱相互作用的评价上取得了丰硕的成果.在各类新型的泛函中,对于氢键结合能的计算和一些弱长程相互作用的描述,M06-2X泛函13表现突出,而且基组不需要过大,就可以得到满意的结果.14本文应用M06-2X/6-31G(d,p)方法对杯[4]吡咯与卤素离子及其离子对的各种组装结构进行全优化,对所得各结构进行频率验证,确保选取的是能量最低的平衡构型,并获得各构型的热力学参数.对确定的结构进行同计算水平的自然键轨道(NBO)10,15分析,以获得形成各组装体系的电荷转移情况及分子间相互作用的类型和强弱,得到电子供体轨道与电子受体轨道之间的二阶微扰稳定化能.这些计算工作均采用Gaussian 09程序包16完成.
分子间弱相互作用的可视化方法是揭示超分子体系本质的重要工具.由结构优化时产生的波函数文件,依据杨伟涛课题组提出的弱相互作用可视化研究方法,17计算了主体杯[4]吡咯与各离子之间在空间上各点的约化密度梯度(RDG)函数和sign(λ2(r))ρ(r)函数,使用 Multiwfn 程序18得到RDG等值面图.
3 结果与讨论
3.1 杯[4]吡咯与卤素离子X-(X-=F-,Cl-,Br-)的1:1复合物
杯[4]吡咯识别卤素阴离子的研究已有诸多报道,6,19且对杯[4]吡咯-卤素阴离子的构型、结合能、识别作用力进行了系统的阐述.在此基础上,本文应用M06-2X/6-31G(d,p)方法,结合先进的分子间相互作用图形显示工具,对杯[4]吡咯与卤素阴离子的作用本质进行了比较研究.
自由状态的杯[4]吡咯最稳定构象为1,3交替式,在1:1卤素离子作用体系中,主体分子由自由状态下的1,3交替构型变成了复合状态下的锥式构型.由图2可见,卤素离子位于头部的中心位置,呈现一种高度对称的构型(C4v),且主体分子上的N―H键长也有不同程度的增长,由自由状态下的0.1011 nm(CP)分别增长到 0.1035 nm(CPF)、0.1022 nm(CPCl)、0.1021 nm(CPBr).可见复合过程会对主体分子上的部分键长有影响,影响幅度随着卤素非金属性的减弱而减小.主体与客体上的N―H…X距离随着卤素离子半径的增大而增大,由0.1666 nm增大到0.2387 nm,且距离都小于相应分子的范德华作用半径,说明存在着明显的相互作用力.N―H…X的键角都在170°左右,满足氢键作用力的形成,因此相互作用力主要是氢键作用力,并且作用力大小也是随着卤素的非金属性的减弱而变小.6,19从表1包合过程能量的变化可以看出,F-包合相互作用力最强,Cl-、Br-作用力大幅低于F-,两者的能量变化相近且Br-出现了逆转,并没有呈现相应的卤素递变规律.这是因为文献6,19中主要运用B3LYP方法,主要考虑氢键作用力,而M06-2X方法考虑了更多的弱相互作用力.12,13虽然Cl-、Br-氢键作用力随着半径增大在递减,而一些弱的远程相互作用,如范德华力、静电力却随着离子半径的增大而增强,此时影响包合作用的过程不仅主要是氢键力,范德华力等分子间的长程相互作用力影响也很大.加上Cl-、Br-组装体系中,两者本身能量变化相差不大,易出现上述能量逆转情况.

图2 杯[4]吡咯与卤素阴离子1:1复合物的优化结构Fig.2 Optimized structures of 1:1 complexes of calix[4]pyrrole and halide anions
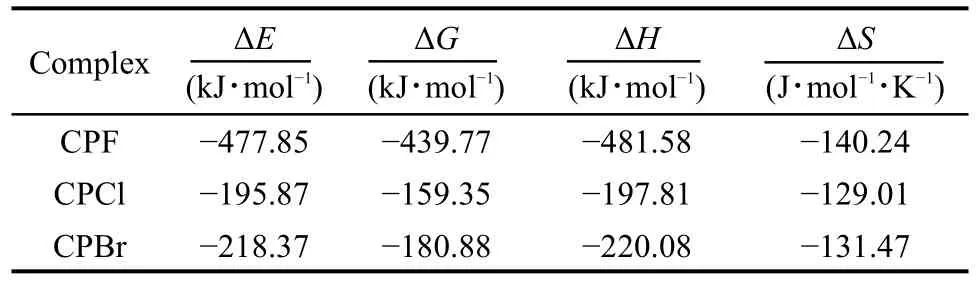
表1 杯[4]吡咯与卤素离子X-(X-=F-,Cl-,Br-)1:1复合物的能量和热力学参数(298 K)Table 1 Calculated energies and thermochemistry parameters(298 K)of calix[4]pyrrole and halide ions X-(X-=F-,Cl-,Br-)1:1 complexes
3.1.1 自然键轨道NBO分析

3.1.2 Multiwfn拓扑及可视化弱相互作用分析
利用Gaussian 09产生的波函数文件,借助Multiwfn软件进行电子密度的拓扑分析,并绘制轨道波函散点图及等值面图,可清楚直观地展示出分子组装体系中的成键情况及弱相互受力作用.
实空间函数(以三维空间坐标为变量的函数)的拓扑分析主要是指获取临界点,并获取连接临界点的拓扑路径;其电子密度分布的拓扑性质取决于电子密度梯度▽ρ(r)和Laplace量▽2ρ(r).临界点(CP)是指函数梯度的模为0的点,可分为四类,以坐标(3,X)表示.实空间函数的Hessian矩阵(3×3的二阶导数矩阵)的本征值个数为3,并且满足▽2ρ(r)=λ1+λ2+λ3,假设其中有M个正值和N个负值,则X=M-N.(3,-3)对应函数的局部极大点通常出现在离原子核很近的位置.(3,-1)对应函数的二阶鞍点,通常出现在存在相互作用的两个原子之间,也被称为键临界点(BCP).(3,+1)对应函数一阶鞍点,如同势能面上的过渡态,通常出现在环体系平面中.(3,+3)则对应函数的局部极大点,通常出现在笼状体系中.从杯[4]吡咯与卤素离子的拓扑成键关系图3中可以看出,卤素离子和吡咯环上与氮相连的氢原子产生了键的临界点,说明其原子之间存在相互作用力,通过N―H结构产生作用力使组装体系达到稳定态的包合,并且可以看出三种不同卤素离子的拓扑成键关系类型,也是一致的.另外成键之后形成的拓扑路径也构成了一个环状体系,与吡咯环一样,存在着一阶鞍点.结合表3中的电子密度及Laplace值可以看出,ρ(r)在0.020-0.045 a.u.之间,▽2ρ(r)处于0.049-0.150 a.u.左右.说明N―H键之间存在着氢卤键作用,且氢卤键作用都是随着卤素电负性的关系呈现着相应的递变规律,F-作用较强,Cl-、Br-依次减弱.并且每个卤素离子与吡咯主体形成的4个键径作用大小受力均等,印证了其结构高度对称规整.
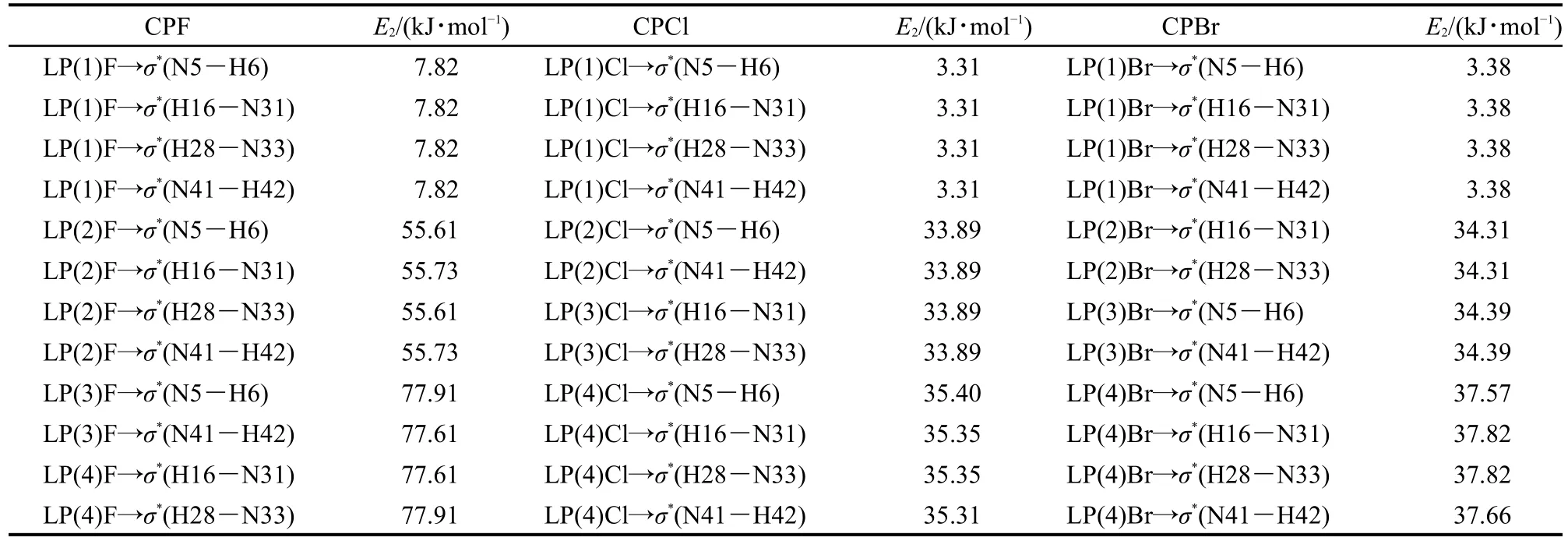
表2 杯[4]吡咯与卤素离子X-1:1复合物NBO结果分析Table 2 NBO analysis of 1:1 complexes of calix[4]pyrrole and halide ions X-
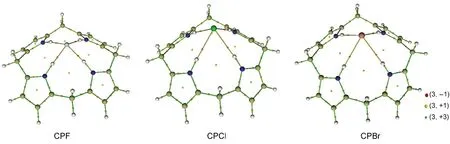
图3 杯[4]吡咯与卤素离子X-拓扑成键关系Fig.3 Topological bonding relationship of calix[4]pyrrole and halide ions X-
为了区分弱相互作用所涉及的区域,定义一个实空间函数,使其数值能够区分开体系中具有不同特征的区域,此方法使用的是约化密度梯度函数(RDG).弱相互作用的临界点ρ(r)是衡量相互作用强度的重要指标之一,其数值和键的强度存在正相关性.sign(λ2(r))函数是电子密度Hessian矩阵的第二大本征值λ2的符号,可以用来反映键的类型.在sign(λ2(r))<0区域其主要是强吸引作用包括氢键、强卤键等;sign(λ2(r))≈0符合的是范德华作用区域,由于区域内电子密度很小,符号较为不稳定,所以可正可负;sign(λ2(r))>0对应于在环、笼中出现的较强位阻,包括原子间的互斥效应.将sign(λ2(r))函数和ρ(r)相乘而得的sign(λ2(r))ρ(r)函数投影到RDG等值面上,则弱相互作用的类型、位置、强度都能直观地显现出来,如图4所示.由RDGvssign(λ2(r))ρ(r)的散点图及填色等值面可以看出,F-形成的体系在sign(λ2(r))<0区域内具有一个很负的扇形突起峰,其作用力为F-与N―H之间形成的氢键和强卤键作用.近0处是一个很窄的针尖峰,主要为原子间的范德华作用.sign(λ2(r))>0区域有个小三角峰对应,则主要是吡咯环中产生的位阻效应.Cl-、Br-体系在三个区域内也具有类似的峰形,其扇形突起峰出现的位置相近,但没有F-位置负,可见Cl-、Br-体系形成的氢键作用力大小相近,但比F-体系弱.另外Cl-、Br-体系与N―H之间还存在着范德华力及位阻效应,其中Cl-位阻效应比Br-要大,使得其稳定包合状态有所降低,这也解释了表1所示Cl-、Br-包合能量不成规律,出现逆转的原因.
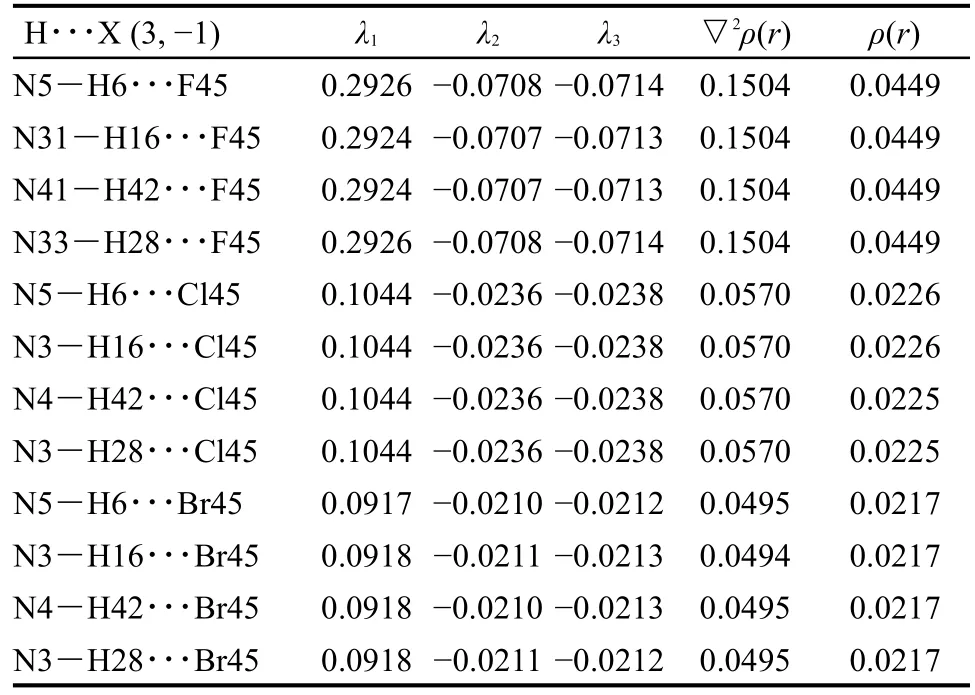
表3 杯[4]吡咯与卤素离子X-的相关电子密度及Hessian矩阵部分参数Table 3 Correlative parameters of electron density and Hessian matrix of calix[4]pyrrole and halogen ions X-

图4 RDG vs sign(λ2(r))ρ(r)的散点图(上)和填色等值面图(下)Fig.4 RDG vs sign(λ2(r))ρ(r)scatter plot(top)and fill isosurfaces(below)
3.1.3 杯[4]吡咯与卤素离子X-的2:1复合物
卤素离子与主体杯[4]吡咯1:1的组装体系呈现的是四方锥式对称(C4v)的包合构型,使得卤素离子能够与环上的每个吡咯单元形成稳定均等的相互作用力,从而使整个体系趋于包合的稳定状态.然而1:1复合物中的卤素离子仍具有复合主体分子的可能,即形成2:1复合体系.在1:1复合物的顶端头碰头加上一个锥式杯[4]吡咯结构,构建主-客体的2:1复合体系(CPXCP),在M06-2X/6-31G(d,p)水平上优化结构如图5所示.
从图5可以看出,在2:1的组装体系中,两个主体分子依然呈现的是锥式构型,但CPClCP和CPBrCP中的第二个主体分子出现了部分扭曲变形.且两主体分子之间头头相对,两端N―H错位排布形成“点状花环”型,这有利于减弱两个主体分子间互斥效应,卤素离子位于两主体分子之间,与两端都能产生非键作用力.F-强大的电负性能够同时对两边的主体产生强烈的束缚作用,主体的构型也就相对稳定.Cl-、Br-由于电负性的大幅减弱,使得氢键作用减弱,长程作用力大幅增加,主体间的静电作用及互斥效应使得组装包合构型扭曲偏转程度变大.对相关键长变化的分析,发现卤素离子与两端形成的键长也趋于平均化,使两边主体受力作用均衡,从而达到一个稳定状态.从表4中包合能量的变化可以看出增加主体分子后,体系扩大,包合能都有一个大幅度的增加,这也有助于提高包合物的稳定性,对于单个卤素离子还是倾向此类组装包合.
3.2 杯[4]吡咯与X--NH4+离子对的包合
杯[4]吡咯中由四个吡咯环围成了一个富电子的空穴,能够与阳离子通过阳离子-π电荷转移作用形成包合物.20在M06-2X/6-31G(d,p)水平上,得到了两种包合物,NH4CP1和NH4CP2(见图1).NH4CP1的能量比NH4CP2的能量低5.13 kJ·mol-1,且在NH4CP1中,N的一个N―H键指向杯[4]吡咯的锥形顶部,更有利于与卤素阴离子形成离子配对作用.在优化所得的离子对包合体系中,N与杯[4]吡咯之间均采取这种作用方式.

图5 杯[4]吡咯与卤素阴离子2:1复合物的优化结构Fig.5 Optimized structures of 2:1 complexes of calix[4]pyrrole and halide anions

表4 杯[4]吡咯与卤素离子X-2:1体系的能量及热力学参数(298 K)Table 4 Calculated energies and thermochemistry parameters(298 K)of 2:1 complexes calix[4]pyrrole and halide ions X-
3.2.1 杯[4]吡咯与X--N离子对的1:1包合
杯[4]吡咯与单个卤素离子能够形成稳定的组装体系,那么与卤素离子-N形成的离子对1:1的包合状态又是怎样的,下面我们对其进行计算研究分析,优化结构和能量变化见图6和表5.
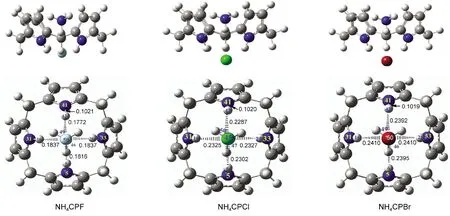
图6 杯[4]吡咯与卤素-铵离子对1:1复合物的优化结构Fig.6 Optimized structures of 1:1 complexes of calix[4]pyrrole and N-X-ion-pairs
表5 杯[4]吡咯与卤素-铵离子对形成1:1复合物的能量及热力学参数(298 K)Table 5 Calculated energies and thermochemistry parameters(298 K)of 1:1 complexes of calix[4]pyrrole and N-X-ion-pairs

表5 杯[4]吡咯与卤素-铵离子对形成1:1复合物的能量及热力学参数(298 K)Table 5 Calculated energies and thermochemistry parameters(298 K)of 1:1 complexes of calix[4]pyrrole and N-X-ion-pairs
Complex NH4CPF NH4CPCl NH4CPBr ΔE(kJ·mol-1)-1033.35-732.06-756.72 ΔG(kJ·mol-1)-953.58-652.33-673.27 ΔH(kJ·mol-1)-1041.06-737.70-763.69 ΔS(J·mol-1·K-1)-293.42-286.32-302.28 ΔY=YNH4CPX-YCP(1,3-alternate)-YX--images/BZ_258_770_927_824_963.png(Y=E,G,H,S)
从图6我们可以看出,杯[4]吡咯与卤素-铵离子对形成的也是一种规整的包合状态且不存在虚频.不同卤素离子与N形成的离子对包合状态都趋于一致,其中N位于杯吡咯空腔内,可以接受吡咯环上的富π电子产生相互作用,卤素位于尾部通过N―H键产生作用力,同时N与卤素离子之间还有强烈的离子间作用力,共同作用力使得离子对包合作用特别的稳定.对相关N―H键及N―H…X键长的变化分析发现也是随着卤素递变规律变化,但键长的变化比单个卤素离子组装体系的变化作用要小,这说明了离子对体系的存在减弱了卤素离子与主体间包括氢键在内的相互作用.从表5的包合能量变化可看出离子对的形成使得稳定包合作用大幅增加,另外卤素-N离子对的组装包合能力大小同样也是没有按照卤素的非金属性呈现相应的递变规律,NH4CPBr的包合能要比NH4CPCl的低24.66 kJ·mol-1.可见非键长程作用力在包合过程中,随着离子半径的增大,是不能忽略的,不能只考虑氢键的主要作用力.
对其拓扑成键关系(图7)分析可以看出卤素离子对包合中,卤素离子分别与吡咯主体上的四个N―H形成键径关系,同时卤素与N之间也存在着键径关系,主要为离子键作用.N除了与卤素之间的离子键作用外,还与主体分子上的部分原子之间存在键径关系,说明之间有长程作用力的存在.网状的键径关系对吡咯环包合构象起着支撑作用,使得整个主客体组装包合更加稳定,大大提高了对离子的识别作用.从表7中的电子密度ρ(r)、Laplace值可以看到离子对中卤素单体离子与主体作用依然随卤素性质变化呈现着递变规律,但有一定的减弱,其中 F-较明显,Cl-、Br-影响很小.卤素与N作用在F-体系中键径作用较大,Cl-、Br-中作用依次减弱.所以在离子对中,其包合作用之所以较稳定,不是通过提高单体离子与主体的作用,而是主体与单体、离子对之间两两相互作用力交织在一起,形成了一个组合体,从而大大提高了体系的稳定.
表6 杯[4]吡咯与N-X-离子对包合物的NBO分析结果Table 6 NBO analysis of calix[4]pyrrole and N-X-ion-pairs

表6 杯[4]吡咯与N-X-离子对包合物的NBO分析结果Table 6 NBO analysis of calix[4]pyrrole and N-X-ion-pairs
Atomic numeral orders are found in Fig.6.
NH4CPF E2/(kJ·mol-1)LP(1)F→σ*(N5―H6)LP(1)F→σ*(H16―N31)LP(1)F→σ*(H28―N33)LP(1)F→σ*(N41―H42)LP(2)F→σ*(H16―N31)LP(2)F→σ*H28―N33 LP(3)F→σ*(N5―H6)LP(3)F→σ*(N41―H42)LP(4)F→σ*(N5―H6)LP(4)F→σ*(H16―N31)LP(4)F→σ*(H28―N33)LP(4)F→σ*(N41―H42)LP(1)F→σ*(H46―N49)LP(4)F→σ*(H46―N49)20.46 17.07 17.07 19.66 46.82 46.74 48.20 60.50 6.07 6.36 6.19 13.18 14.39 232.13 NH4CPCl E2/(kJ·mol-1)LP(1)Cl→σ*(N5―H6)LP(1)Cl→σ*(H16―N31)LP(1)Cl→σ*(H28―N33)LP(1)Cl→σ*(N41―H42)LP(2)Cl→σ*(H16―N31)LP(2)Cl→σ*(H28―N33)LP(3)Cl→σ*(N5―H6)LP(3)Cl→σ*(N41―H42)LP(4)Cl→σ*(N5―H6)LP(4)Cl→σ*(H16―N31)LP(4)Cl→σ*(H28―N33)LP(4)Cl→σ*(N41―H42)LP(4)Cl→σ*(H47―N50)6.57 5.81 5.77 6.52 36.82 37.82 46.57 33.60 8.58 13.26 11.92 24.35 40.54 NH4CPBr E2/(kJ·mol-1)LP(1)Br→σ*(N5―H6)LP(1)Br→σ*(H16―N31)LP(1)Br→σ*(H28―N33)LP(1)Br→σ*(N41―H42)LP(2)Br→σ*(H16―N31)LP(2)Br→σ*(H28―N33)LP(3)Br→σ*(N5―H6)LP(3)Br→σ*(N41―H42)LP(4)Br→σ*(N5―H6)LP(4)Br→σ*(H16―N31)LP(4)Br→σ*(H28―N33)LP(4)Br→σ*(N41―H42)LP(4)Br→σ*(H46―N49)6.32 5.94 5.90 6.19 41.46 41.67 46.53 40.12 16.32 18.20 17.90 24.23 39.83

图7 杯[4]吡咯与N-X-离子对的拓扑成键关系Fig.7 Topological bonding relationship of calix[4]pyrrole and N-X-ion-pairs
表7 杯[4]吡咯与N-X-离子对的相关电子密度及Hessian矩阵部分参数Table 7 Correlative parameters of electron density and Hessian matrix of calix[4]pyrrole and N-X-ion-pairs

表7 杯[4]吡咯与N-X-离子对的相关电子密度及Hessian矩阵部分参数Table 7 Correlative parameters of electron density and Hessian matrix of calix[4]pyrrole and N-X-ion-pairs
all units in a.u.
λ1λ2λ3 H···X(3,-1)N5―H6···F50 N31―H16···F50 N41―H42···F50 N33―H28···F50 N49―H46···F50 N5―H6···Cl45 N31―H16···Cl45 N41―H42···Cl45 N33―H28···Cl45 N50―H47···Cl45 N5―H6···Br50 N31―H16···Br50 N41―H42···Br50 N33―H28···Br50 N49―H46···Br50 0.2003 0.1904 0.2214 0.1902 0.3998 0.1041 0.0994 0.1054 0.0992 0.0683 0.0950 0.0924 0.0947 0.0923 0.0586-0.0462-0.0438-0.0515-0.0437-0.1170-0.0225-0.0211-0.0231-0.0210-0.0150-0.0213-0.0205-0.0213-0.0204-0.0131-0.0419-0.0398-0.0483-0.0398-0.1171-0.0205-0.0192-0.0214-0.0191-0.0150-0.0196-0.0189-0.0199-0.0188-0.0132▽2ρ(r)0.1122 0.1068 0.1217 0.1067 0.1657 0.0612 0.0591 0.0609 0.0591 0.0382 0.0542 0.0530 0.0534 0.0530 0.0324 ρ(r)0.0329 0.0316 0.0357 0.0316 0.0622 0.0212 0.0203 0.0217 0.0202 0.0160 0.0214 0.0208 0.0215 0.0208 0.0152
对卤素离子对的散点图及填色等值面(图8)进行分析可以得到,在离子对的包合过程中,离子间作用力的存在减弱了卤素与主体形成的氢键作用,扇形峰都有一定的正移,其中N-F-影响最大.离子对体系的存在使得主客体之间包括范德华作用在内的长程作用力大幅增加,其中Cl-、Br-体系增加程度较大,sign(λ2)≈0处出现了密集的针尖峰,位阻效应则主要出现在N与主体之间及吡咯环内.
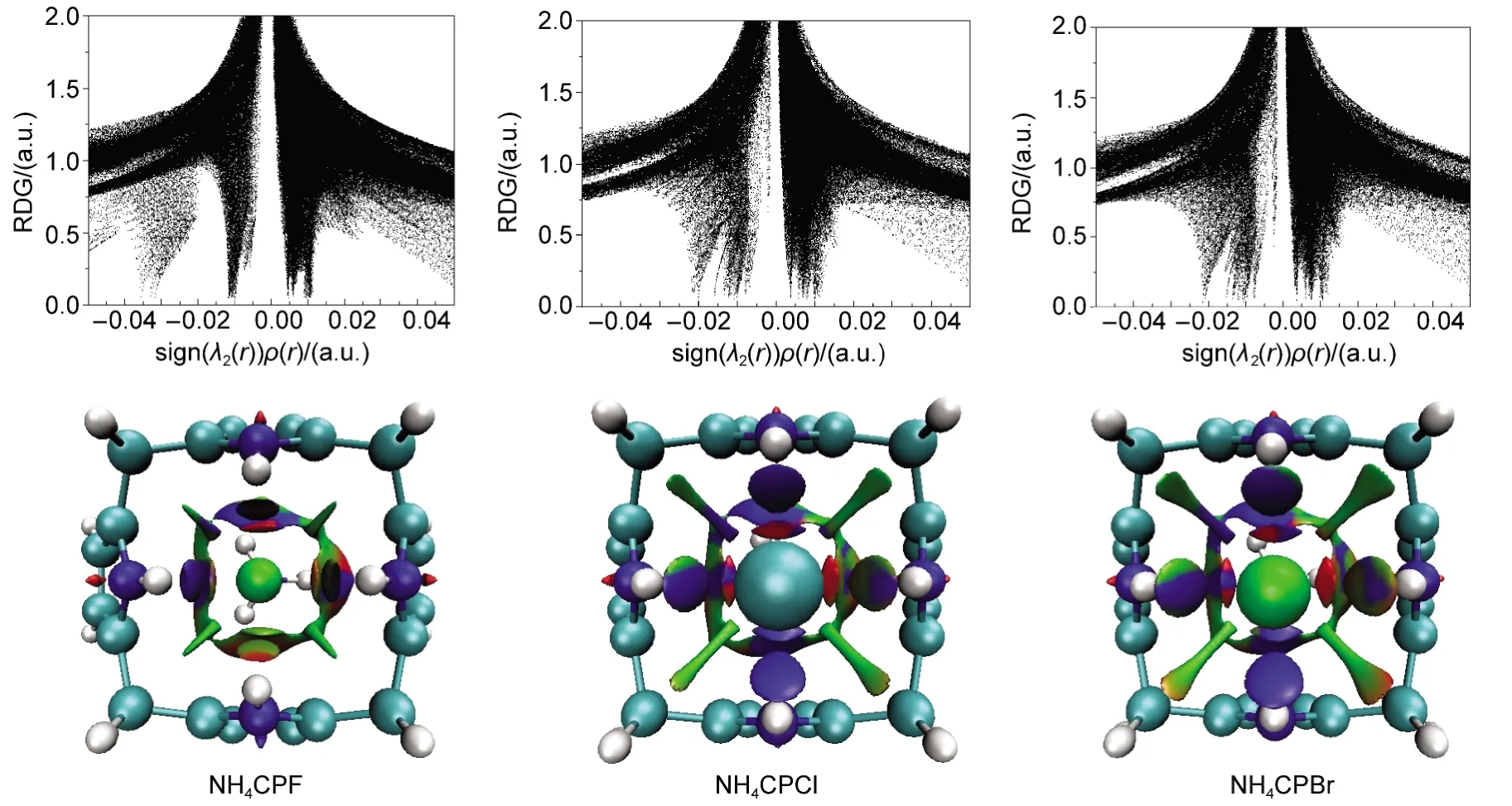
图8 RDG vs sign(λ2(r))ρ(r)的散点图(上)和填色等值面图(下)Fig.8 RDG vs sign(λ2(r))ρ(r)scatter plot(top)and fill isosurfaces(below)
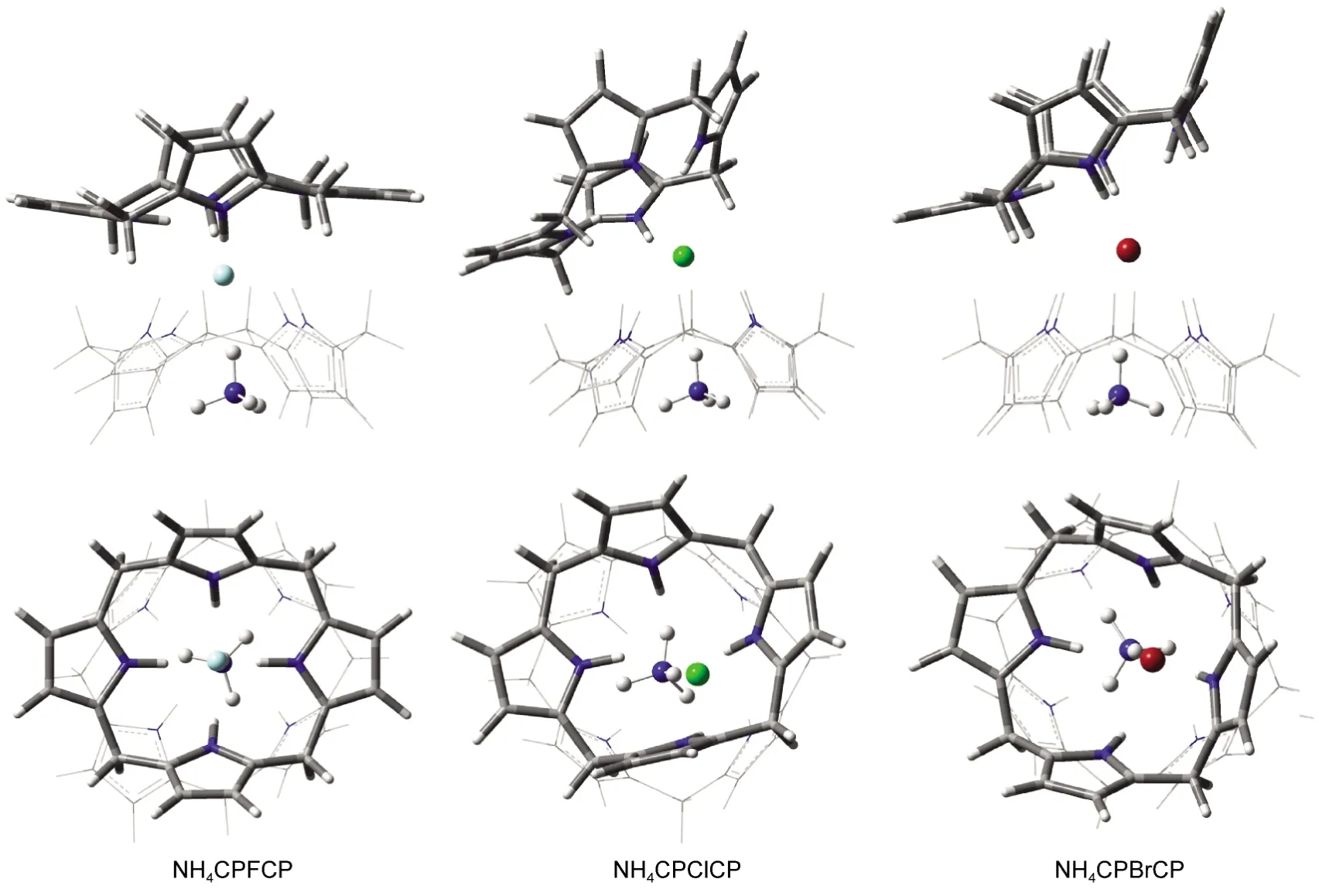
图9 杯[4]吡咯与N-X-离子对的2:1复合物的优化结构Fig.9 Optimized structures of 2:1 complexes of calix[4]pyrrole and N-X-ion-pairs
我们还尝试了杯[4]吡咯与离子对的另一种作用方式,见图1中的CPXNH4的构型.在这个结构中主体分子与阴离子直接作用,阳离子只与阴离子作用,体系中同样存在离子配对作用.以CPFNH4和NH4CPF比较可见,虽然前者阴、阳离子的距离缩短,离子配对作用增强,却导致阴离子与主体分子的作用减弱,使整个结构趋于松散、扭曲.从结合能上看,CPFNH4比NH4CPF小56.87 kJ·mol-1,因此前者的稳定性明显弱于后者.CPClNH4和CPBrNH4的情况也是如此,本文对CPXNH4构型不再赘述.
3.2.2 杯[4]吡咯与X--N离子对的2:1包合
由计算结果可知离子对主客体2:1的包合(图9),同样延续了2:1的单个离子的包合构型,都能呈现很好的错位“点状花环”构型,两个主体头头相对,卤素离子夹在中间,N离子位于一侧主体空腔内.其中一侧无N主体构型的扭曲偏转程度进一步的加大,与N离子包合的这端主体构型相对规整些,这得益于N离子位于主体空腔内也可以对周围主体上的原子产生静电力的束缚作用,从而减弱了空腔的柔性作用,构型变化就受到了一定的限制.没有N离子的这端主体,由于受到卤素离子作用的减弱,形成的键力不强,加上主体间互斥效应的存在使得主体各处受力不均衡,构型不稳定,很容易发生偏转和变形.另外不同卤素离子的电负性的不同,产生的作用力对主体的影响程度也就不同,构型的变化程度也就有了差异.对比表8和表5能量的变化发现2:1的离子对体系其包合能量与1:1体系的包合能量的变化不大,增加的幅度很小,所以其包合构型更倾向1:1的体系.对2:1的组装体系的另一种构型为图1中的CPXNH4CP,该构型虽然能得到优化结果,但由于阳离子与杯[4]吡咯空腔的作用非常弱,导致这种构型非常松散,包合能很小,因而不再考虑该类构型.
表8 杯[4]吡咯与N-X-离子对的2:1体系的能量及热力学参数(298 K)Table 8 Calculated energies and thermochemistry parameters(298 K)of 2:1 complexes of calix[4]pyrrole and N-X-ion-pairs

表8 杯[4]吡咯与N-X-离子对的2:1体系的能量及热力学参数(298 K)Table 8 Calculated energies and thermochemistry parameters(298 K)of 2:1 complexes of calix[4]pyrrole and N-X-ion-pairs
Complex NH4CPFCP NH4CPClCP NH4CPBrCP ΔE(kJ·mol-1)-1099.82-773.94-823.33 ΔG(kJ·mol-1)-947.30-633.92-678.49 ΔH(kJ·mol-1)-1108.18-776.97-828.23 ΔS(J·mol-1·K-1)-539.61-479.79-502.25 ΔY=YNH4CPXCP-2YCP(1,3-alternate)-YX--YNH+4(Y=E,G,H,S)
4 结论
在M06-2X/6-31G(d,p)计算水平上,对杯[4]吡咯与卤素阴离子以及卤素-铵离子对的相互作用进行了系统研究.尤其是对杯[4]吡咯与卤素阴离子1:1复合物(CPX)及其离子对的1:1复合物(NH4CPX),从结构、能量、NBO、拓扑结构方面进行了详细地比较分析.结果显示,杯[4]吡咯在包合单个卤素离子时,卤素其强烈的电负性可以与主体上的N―H产生键的作用力,主要为卤键、氢键作用.卤素离子位于主体的头部,这使得其与主体间的位阻很小,可以同时与两侧的主体产生作用.卤素电负性越强,其作用力越强,对两侧的主体束缚作用越强,主体间的弱相互作用力对主体的组装构型影响就越小,形成的主客体组装体系也就倾向于2:1体系.对于电负性小的离子,其长程作用力对主体构型的影响增大,此时构型也就由氢键、静电作用及范德华等作用力共同影响,形成大体系时各处受力不均衡其构型就会发生一些倾斜偏转.卤素离子对的组装体系延续了单个卤素离子的作用方式,卤素离子还是位于头部,阳离子则位于主体空腔内.一方面阳离子与卤素阴离子之间有着强烈的离子间作用力,另一方面其离子对也同时与相邻的主体作用,共同作用力的存在使得主体被离子对牢牢束缚.但离子间作用力的存在却减弱了单个卤素离子与主体上的N―H作用,其对主体作用的减弱,使得束缚2个主体时构型就不稳定,所以卤素离子对的组装包合更倾向于1:1的主客体体系.
(1) Gale,P.A.;Anzenbacher,P.;Sessler,J.L.Coord.Chem.Rev.2001,222,57.doi:10.1016/S0010-8545(01)00346-0
(2)Allen,W.E.;Gale,P.A.;Brown,C.T.;Lynch,V.M.;Sessler,J.L.J.Am.Chem.Soc.1996,118,12471.doi:10.1021/ja9632217
(3) Rambo,B.M.;Sessler,J.L.Chem.Eur.J.2011,17,4946.doi:10.1002/chem.v17.18
(4) Cafeo,G.;Carbotti,G.;Cuzzola,A.;Fabbi,M.;Ferrini,S.;Kohnke,F.H.;Papanikolaou,G.;Plutino,M.R.;Rosano,C.;White,A.J.P.J.Am.Chem.Soc.2013,135,2514.
(5) Blas,J.R.;Marquez,M.;Sessler,J.L.;Luque,F.J.;Orozco,M.J.Am.Chem.Soc.2002,124,12796.doi:10.1021/ja020318m
(6)Wu,Y.D.;Wang,D.F.;Sessler,J.L.J.Org.Chem.2001,66,3739.doi:10.1021/jo0016273
(7) Pichierri,F.J.Mol.Struct.2002,581,117.
(8) Wintergerst,M.P.;Levitskaia,T.G.;Moyer,B.A.;Sessler,J.L.;Delmau,L.H.J.Am.Chem.Soc.2008,130,4129.
(9) Kriz,J.;Dybal,J.;Makrlik,E.;Sedlakova,Z.J.Chem.Phys.2012,400,19.
(10) Xia,Y.;Wang,X.;Zhang,Y.;Luo,B.;Liu,Y.Journal of Molecular Modeling2012,18(6),2291.
(11) Blas,J.R.;Marquez,M.;Sessler,J.L.;Luque,F.J.;Orozco,M.Chem.Eur.J.2007,13,1108.
(12) Zhao,Y.;Truhlar,D.G.Accounts Chem.Res.2008,41,157.doi:10.1021/ar700111a
(13) Zhao,Y.;Truhlar,D.G.Theor.Chem.Acc.2008,120,215.doi:10.1007/s00214-007-0310-x
(14) Sun,T.;Wang,Y.B.Acta Phys.-Chim.Sin.2011,27(11),2553.[孙 涛,王一波.物理化学学报,2011,27(11),2553.]doi:10.3866/PKU.WHXB20111017
(15) Weinhold,F.;Schleyer,P.v.R.;Clark,T.;Gasteiger,J.;Kollman,P.A.;SchaeferI,H.F.,III;Schreiner,P.R.Encyclopedia of Computational Chemistry;John Wiley&Sons:Chichester,UK,1998;Vol.3,pp 1792-1811.
(16) Frish,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,Revision B1;Gaussian Inc.:Wallingford,CT,2010.
(17) Johnson,E.R.;Keinan,S.;Mori-Sanchez,P.;Contreras-Garcia,J.;Cohen,A.J.;Yang,W.J.Am.Chem.Soc.2010,132,6498.
(18) Lu,T.;Chen,F.J.Comput.Chem.2012,33,580.
(19)Chen,P.Q.;Sun,H.W.;Chen,L.;Shen,R.X.;Yuan,M.X.;Lai,C.M.Chem.J.Chin.Univ.2004,25(12),2290.[陈沛全,孙宏伟,陈 兰,沈荣欣,袁满雪,赖城明.高等学校化学学报,2004,25(12),2290.]
(20) Liu,K.;Guo,Y.;Xu,J.;Shao,S.J.;Jiang,S.X.Chinese Chemical Letters2006,17(3),387.
